Sex and Gender Identification and Implications for Disability Evaluation (2024)
Chapter: 7 Care for Individuals with Variations in Sex Traits
7
Care for Individuals with Variations in Sex Traits
Society and biology generally view sex as a “binary.” Typically, a baby’s sex as “girl” or “boy” is identified in the delivery room immediately after birth based on the appearance of the baby’s external genitalia. Increasingly, the baby’s sex is being ascertained through prenatal ultrasound imaging and/or noninvasive prenatal screening studies. Sometimes, however, the appearance of the external genitalia does not inform determination of the infant’s biological sex because of unconventional or atypical appearance of the external genital anatomy.1 In these cases, ascertaining or labeling a child’s sex may be challenging. Diverse and heterogeneous etiologies can affect development of the reproductive tract, resulting in atypical genital development.
Atypical genitalia are estimated to occur in approximately 1 in 4,500 live births. Several terms are used interchangeably to describe this condition (Hughes, 2007). These terms include “ambiguous genitalia,” “variations of sex development,” “variants of sex traits” (VSTs), “variations in sex characteristics,” “intersex,” and “differences of sex development” (DSD). For the purposes of this report, the committee uses the term “variations in sex
___________________
1 Infants with atypical external genitalia may have a “phallus,” the term used to describe both a clitoris and a penis. In the usual situation, 46,XY fetuses secrete testosterone from their testes, causing the phallus to develop into a penis, whereas in 46,XX fetuses, the absence of testosterone causes the phallus to develop into a clitoris. Where the genitalia are ambiguous, the phallus may not have fully developed. Infants with variations in sex traits may also have the urethral meatus on the perineum and nonpalpable gonads. Other infants may have an asymmetric appearance of the external genitalia, with an ostensibly rugated scrotum and palpable gonad on one side and an apparent labia majora and nonpalpable gonad on the other side.
traits,” or “VSTs,” when describing people with atypical genital development and other clinical presentations as described in this chapter.2
In some instances, the clinical presentation of a VST condition occurs beyond infancy, during childhood, adolescence, or adulthood. In total, estimates of the percentage of the population born with VSTs vary from 0.05 to 1.7 percent, depending on the definition used for VST (e.g., restricting the definition to include only those with atypical genitalia or taking a broader approach to include other differences in characteristics or reproductive anatomy) and the type of study conducted (medical or general population study) (Blackless et al., 2000). Overall, the statistics for the occurrence of VST are possibly inaccurate, given the difficulty patients with VSTs face in accessing knowledgeable medical providers and the variability in medical documentation.
Much variability exists in clinical presentations and precise etiologies. Sex determination and sex development involve complex sequential biologic networks with carefully orchestrated time-based interactions between specific molecular signals and hormones, and cross-talk between these signaling pathways. Hence, clinical features, age at presentation, diagnostic evaluations, appropriate health care interventions, clinical course, and specific etiologies are quite variable. Given the global perception of biological sex as perpetually binary, affected individuals, clinicians, parents or other caregivers, and family members often struggle when confronted with these conditions. While appropriate care may include hormonal treatment (or, in some cases, surgical treatment), behavioral/mental/psychosocial support, guidance, and counseling are essential for children, adolescents, emerging adults, and adults with VSTs and their caregivers and family members.
This chapter provides an overview of many of the conditions included under the VST rubric, describes common care management for people with VSTs, reviews potential hormonal and surgical interventions, and examines psychosocial/mental health/behavioral health care for this population.
___________________
2 The committee chose to use the term “variations in sex traits” (VSTs) for this report to describe individuals with variations in development of the reproductive system (sex determination and sex development). The term “differences of sex development” (DSD) is commonly used in medical records and in the medical literature. However, some patients and clinicians consider the term “DSD” to be inaccurate and distasteful. “Intersex” is another commonly used term, but some individuals take issue with the notion that their reproductive anatomy falls between the binary and question this terminology. “Variations in sex traits,” therefore, is intended to encompass all variations in reproductive tract development while being attentive to patient lived experience. However, the committee acknowledges that stakeholders have different and varied opinions on appropriate terminology, and, as is true for other terminology in this report, this terminology is likely to evolve over time. Table 2-3 in Chapter 2 displays a range of terms that may be present in medical records to describe VSTs, including some terms that have fallen out of popular use and may be considered offensive (including “pseudohermaphroditism” and “hermaphroditism”); however, these terms may still be found in older medical records.
Long-term health concerns and considerations are reviewed. While this chapter refers to guidelines and common care practices, it is not within the charge of this committee to recommend preferred practices; rather the committee’s charge in response to the statement of task is to present the range of care practices that may exist and therefore may be seen in medical records submitted to the Social Security Administration (SSA) as part of a disability application.
CLASSIFICATIONS OF VARIATIONS IN SEX TRAITS
In 2005, recognizing inconsistences in terminology and health care among people with VSTs, an expert consensus meeting was held in Chicago to examine the vocabulary and health care for individuals with VSTs from a broad perspective (Hughes et al., 2006). The participating experts reviewed available patient outcomes, medical treatments, psychosocial management, potential surgical considerations, and gender concerns. Discussions focused on improving diagnostic approaches, expanding patient and family participation in medical decision making, involving multidisciplinary health care teams, and advancing medical management strategies. This conference represented a milestone in improving health care for individuals with VSTs, especially the introduction of a multidisciplinary team approach.
The consensus meeting concluded that rigid algorithms and guidelines were inappropriate because of the need for individualized treatment, and that additional outcome data were essential to developing future clinical guidelines (Lee et al., 2016). The “Consensus Statement on Management of Intersex Disorders” surfaced from this meeting, which proposed a classification system with three major categories to assist with the VST diagnostic process. Outlined in Box 7-1, these categories are sex chromosome DSD; 46,XX DSD; and 46,XY DSD (Hughes et al., 2006). Importantly, for some VST conditions, the manifestations are limited to the reproductive system, whereas other conditions may have additional features, such as renal agenesis, hearing loss, and developmental delay (Cox et al., 2014). Box 7-1 defines the three major DSD categories, with subcategories underneath.
Critiques of the 2006 Consensus Statement3 prodded stakeholders to continue to modify the terminology and classification of VSTs to better categorize the underlying genetic etiologies (Aaronson and Aaronson, 2010;
___________________
3 Criticisms of the DSD classification system presented in Box 7-1 are that it does not achieve its goals of bringing clarity and precision to VST diagnosis or care, and that terms used (particularly “disorder” or other pathologizing terms) may medicalize nonpathological variations. In addition, VST scholars and organizations have criticized the 2006 consensus statement process for failing to adequately involve persons with lived experience, who, research demonstrates, have different and varied opinions on appropriate terminology and categorization (Lundberg et al., 2018).
BOX 7-1
Consensus Statement on Management of Intersex Disorders, 2006
Sex Chromosome DSD: A category of VSTs that includes any condition in which there is an atypical number/arrangement of the sex chromosomes.
- 45,X (Turner syndrome and variants)
- 47,XXY (Klinefelter syndrome and variants)
- 45,X/46,XY (mixed gonadal dysgenesis, ovotesticular DSD)
- 46,XX/46,XY (chimeric, ovotesticular DSD)
46,XY DSD: Children born with XY chromosomes (46,XY) usually develop male physical sex characteristics. However, some have underdeveloped gonads or cannot produce or respond to sex hormones to develop the typical male physical characteristics.
- Disorders of gonadal (testicular) development
- Disorders in androgen synthesis or action
- Other
46,XX DSD: Children born with two X chromosomes (46,XX) usually develop female physical sex characteristics. However, some were exposed before birth to excess male sex hormones that led to genitals that appear atypical.
- Disorders of gonadal (ovarian) development
- Androgen excess
- Other
SOURCE: Adapted from Hughes et al. (2006).
Acién and Acién, 2020; Davies et al., 2011; Lee et al., 2016). This committee does not offer commentary on the appropriate categorization of VSTs; however, medical records since 2006 often use the classification system displayed in Box 7-1 when describing patients with VSTs because this classification provides a system for organizing the many different VST conditions (Pasterski et al., 2010). Terminology used prior to 2006 was often inconsistent and mainly descriptive; therefore, older medical records may not use the 2006 terminology.
The following sections examine the VSTs within each of the three major categories displayed in Box 7-1. This presentation does not represent an
exhaustive listing of every possible VST, but a description of many common conditions. In the discussion in Chapter 3 of the codes of the International Classification of Diseases and Related Health Problems (ICD), the report presents numerous ICD codes that may be used in medical records to indicate a VST.
Sex Chromosome DSD
Sex chromosome DSD is characterized by sex chromosome aneuploidy (or atypical number/arrangement of the sex chromosomes). Subcategories include Turner syndrome, Klinefelter syndrome, and 47,XYY, as described in the subsections below (Tallaksen et al., 2023). The sex chromosome DSD category also includes individuals with mosaic karyotypes (e.g., 45,X/46,XY; 45,X/46,XY/47,XXX), which are associated with multiple distinct peripheral blood cell lines. This condition is known as mosaicism; people with “mosaic” chromosomes have different chromosome patterns in some cells of the body as a result of random differences in how cells divide while an embryo is growing (McCoy, 2017). Importantly, sex chromosome aneuploidy may not be detected in all cells because of the variable presence of these multiple cell lines.
Turner Syndrome
Turner syndrome is characterized by the absence or structural abnormalities of an X chromosome. The incidence of Turner syndrome is approximately 1 in 2,500 live female births (Gravholt et al., 2023a; Yoon et al., 2023). Common clinical features include short stature, congenital heart disease, horseshoe kidney, webbed neck, and premature ovarian insufficiency. Premature ovarian insufficiency usually presents as complete absence of pubertal development in a pubertal-aged girl and leads to infertility. Girls and women with Turner syndrome have an increased risk for autoimmune disorders, hypertension, type 2 diabetes, lymphedema, chronic ear infections, neurosensory hearing loss, numerous nevi, and osteoporosis (Augoulea et al., 2019; De Sanctis and Khater, 2019; Mitsch et al., 2023; Sandahl et al., 2020). Most girls and women with Turner syndrome have intelligence levels that mirror the spectrum seen in the general population. However, many struggle with educational challenges, developmental delay, and features of autism spectrum disorder (ASD) (Hutaff-Lee et al., 2019). Anxiety, low self-esteem, neurocognitive dysfunction, and poor social skills often occur. External genital development is typical female; median age of diagnosis is 15 years. Girls with Turner syndrome typically present for health care because of short stature, delayed puberty, or infertility (Gravholt et al., 2023a).
Klinefelter Syndrome
Klinefelter syndrome is characterized by the presence of additional X chromosomes in an individual carrying a Y chromosome. The most common karyotype is 47,XXY. Reported prevalence is approximately 1.5 in 1,000 live births (Morris et al., 2008). Median age at diagnosis is 27 years; the presenting complaint is often infertility due to impaired testicular function. Some boys are diagnosed during evaluation for behavioral abnormalities, such as attention-deficit/hyperactivity disorder (ADHD), dyslexia, ASD, or poor school performance. Boys may present with delayed onset of or failure to complete puberty.4 In addition to hypogonadism and infertility, health concerns include neurocognitive dysfunction, anxiety, depression, flat feet (pes planus), gynecomastia, hypotonia, extragonadal germ cell neoplasia, osteopenia, and autoimmune disorders (Foland-Ross et al., 2023; Jordan et al., 2023). Men with Klinefelter syndrome have increased risk for diabetes, thromboembolic events, and cardiovascular disease (Gravholt et al., 2023b).
47,XYY Karyotypes
The phenotypes of patients with 47,XYY karyotypes are poorly characterized, largely because of underdiagnosis. Reported clinical features include tall stature; scoliosis; learning difficulties, including ADHD; and autism. The reported incidence is approximately 1 in 1,000 men (Davis et al., 2020; El-Dahtory and Elsheikha, 2009). Median age at diagnosis is 15 years. In contrast with Klinefelter syndrome, pubertal development tends to be normal. Subfertility is common. Men with 47,XYY karyotypes have increased risk for diabetes, dyslipidemia, asthma, and obstructive lung disease (Riddler et al., 2023). In a longitudinal Danish study, men with 47,XYY had better fertility, increased mortality, and lower socioeconomic status compared with men with Klinefelter syndrome (47,XXY) (Berglund et al., 2020; Ridder et al., 2023).
Among the Million Veteran Program cohort,5 approximately 1 in 370 men were found to have sex chromosome aneuploidy, with an additional X or Y chromosome being present in 145 and 125 per 100,000 males,
___________________
4 It should be noted that “delayed puberty” as described here is not the same thing as puberty delay induced by medications, as described in Chapter 5. Delayed puberty is a medical condition, where puberty happens later than expected or never starts as part of atypical development. Puberty-delaying medications are used to delay puberty in adolescents who seek this intervention as part of gender-affirming care. These medications may also be used for cisgender children to treat precocious puberty (marked by breast development before age 8 or testes growth before age 9).
5 The Veteran’s Health Administration Million Veteran Program is a voluntary population-based study of genetic determinants of various illnesses and health outcomes for individuals who have served in the U.S. military.
respectively (Davis et al., 2023). This prevalence is comparable to population estimates. Thus, men with sex chromosomal aneuploidy, either 47,XXY or 47,XYY, are often underdiagnosed and unrecognized.
45,X/46,XY, Mixed Gonadal Dysgenesis
Individuals with 45,X/46,XY mixed gonadal dysgenesis have a mosaic karyotype consisting of at least two distinct cell lines: 45,X and 46,XY. Development of the internal and external genital structures can vary (Lindhardt Johansen et al., 2012). The external genitalia may range from appearing like those of girls to those of relatively typical-appearing phenotypic boys. Specific clinical features (phenotype) vary. Individuals with this karyotype may have short stature and, like girls with Turner syndrome (discussed later in this chapter), may benefit from growth hormone treatment (Lindhardt Johansen et al., 2012). Based on an older study evaluating neonatal chromosome analyses, many individuals with this karyotype appear as phenotypic males (Hsu, 1989).
Gonadal development is usually aberrant in individuals with mosaic karyotypes, resulting in “streak” (underdeveloped) gonads and/or abnormal testicular development (Acién and Acién, 2020). Typically, one gonad is a streak gonad and the other a dysgenetic testes. In some instances, a gonad is considered to be an “ovotestis” because of the presence of both ovarian and testicular elements, as indicated by the presence of both oogonia and seminiferous tubules. Individuals with 45,X/46,XY karyotypes often present for medical evaluation because of asymmetric external genitalia. Importantly, the risk for gonadal germ cell neoplasia is high in these individuals, necessitating early gonadectomy (Berklite et al., 2019).
Chimeric 46,XX/46,XY
Chimeric 46,XX/46,XY individuals are extremely rare. Phenotypes range from typical girl to typical male appearance. Gonadal histology is also variable and may show ovotestis in which a gonad has both ovarian follicles and seminiferous tubules.
46,XY DSD
The category of 46,XY DSD encompasses anomalous testicular development, defective testosterone synthesis, abnormal cellular response to testosterone (androgen insensitivity disorders), and abnormal synthesis of or response to anti-Müllerian hormone. These disorders occur in both complete and partial forms. The complete forms are more often referred for medical evaluations because of the severity of their symptoms. The appearance of
the external genitalia may be atypical in individuals with aberrant testicular development, defective testosterone synthesis, and androgen insensitivity. The appearance of the external genitalia can range from female to underdeveloped male.
Studies in the 1950s suggested that an infant’s sex could be assigned if the external genitalia and upbringing corresponded to that sex (Money, 1952). Based on this hypothesis, males born with underdevelopment of their external genital structures, such as aphallia, were assigned to female sex for rearing and underwent gonadectomy (Wisniewski et al., 2019; Witchel et al., 2022). Parents were instructed never to share information about the “sex reversal” with anyone, especially the affected individual. Thus, medical records for older individuals may indicate sex reassignment (boy to girl), gonadectomy, and reconstructive surgery of the external genital structures. Indeed, because of past practices regarding secrecy, some affected individuals may be unaware of the full extent of surgical interventions performed during their early childhood.
Clinical observations led to questioning and eventual discarding of this hypothesis. A 2004 study involving the follow-up of genetic males with cloacal exstrophy treated with sex reversal and gonadectomy exposed the importance of prenatal androgen exposure to gender identity; 8 of 14 individuals assigned female sex at birth subsequently transitioned to male (Reiner and Gearhart, 2004). Hence, with the exception of individuals with complete androgen insensitivity syndrome, most 46,XY individuals with underdeveloped male external genitalia are currently raised as male and provided with testosterone replacement therapy as needed (Wisniewski, 2012; Witchel et al., 2022).
Aberrant Testicular Development
Testicular testosterone production results from a series of enzymatic steps occurring within the testes. Genetic variants in these enzymes can interfere with in utero testosterone production, resulting in fetal testosterone deficiency and underdevelopment of the external genitalia. The phenotype of the external genitalia in these 46,XY infants ranges from essentially female to small penis and hypospadias (Domenice et al., 2022). Genetic variants in two specific enzymes—17-beta hydroxysteroid dehydrogenase type 3 (HSD17B3) and 5-alpha reductase type 2 (SRD5A2)—warrant additional discussion because affected infants have atypical genitalia at birth and virilize with the onset of puberty (Bergougnoux et al., 2023). In some areas of the world, infants with HSD17B3 or SRD5A2 variants are raised as girls and transition to a male role during puberty (Imperato-McGinley et al., 1991). However, male assignment at birth often occurs with early identification and diagnosis.
Disorders of Androgen Synthesis or Action
Individuals with androgen insensitivity syndrome (AIS) are born with typical testes that secrete testosterone (Tyutyusheva et al., 2021). However, the cells of these individuals do not respond typically to the testosterone they produce. AIS appears on a spectrum, whereby some individuals have complete AIS (their bodies cannot react to testosterone), and others have partial AIS (their bodies have a reduced response to testosterone). In individuals with complete AIS, the external genitalia appear consistent with typical female genitalia, apart from palpable gonads within the labia. These individuals have a vaginal pouch and lack a uterus. People with partial AIS may be born with undescended or partially descended testes and with various genital differences (e.g., a shallow vaginal opening, a phallus that may be perceived as a large clitoris or a small penis). The prevalence of complete AIS has been estimated to be 1 in 20,000–64,000 XY (karyotypically male) births; the prevalence for partial AIS is unknown (Mendoza and Motos, 2013).
Complete AIS is due most commonly to genetic variants located in the androgen receptor gene (Gottlieb et al., 2012). Typically, individuals with complete AIS are raised as girls. In the past, gonadectomies were performed at young ages. Today, with observations that the risk for gonadal tumor is low and that pubertal testicular testosterone secretion is converted to estrogens (enabling spontaneous pubertal breast development), gonadectomy is often postponed or avoided (Barros et al., 2021; Chaudhry et al., 2017). Some women with complete AIS choose to retain and internally relocate the gonads to improve tumor surveillance imaging (Tack et al., 2018). If gonadectomy is performed, estrogen therapy is essential to preserve bone health (Ko et al., 2017). If gonadectomy was performed in childhood, estrogen treatment will also be needed for breast development (Sultan et al., 2014). Women with complete AIS often perform self-vaginal dilatation to elongate their vaginal pouches to enable sexual intercourse. A uterine transplant, although rare, may enable a person to carry a pregnancy. Individualized health care is essential for patients with AIS, especially those with partial AIS, to accord with individual preferences and phenotypic heterogeneity (Chen et al., 2023).
Disorders of Anti-Müllerian Hormone Synthesis or Action
During fetal male development, the testes secrete anti-Müllerian hormone (AMH) in addition to testosterone. This hormone promotes degradation of the fetal Müllerian ducts. The phenotype of a 46,XY individual with genetic variants in either the AMH gene or the AMH receptor (AMHR2) gene shows typical male phallus; typical placement of the urethra meatus;
and, usually, inguinal (groin) hernia and undescended testes. Internally, uterine tissue is present. This rare autosomal recessive entity is called persistent Müllerian duct syndrome and has been reported in only a few hundred cases in the literature (Brunello and Rey, 2022; Da Aw et al., 2016). Infertility is common because the vas deferens travels through the uterine tissue. However, fatherhood may be achieved by testicular aspiration followed by intracytoplasmic sperm injection (Fang et al., 2021). Rarely, uterine tissue is identified during abdominal surgery in an otherwise typically developed man. Women carrying variants on both alleles have no obvious clinical features.
46,XX DSD
46,XX DSD in Females
In females, the category 46,XX DSD encompasses anomalous ovarian development, disorders of androgen excess, and vaginal or uterine atresia.
Anomalous ovarian development
The list of genetic variants associated with impaired ovarian development and/or function continues to expand. Individuals with these variants have typical female external genitalia at birth; they typically present for evaluation during the adolescent years with delayed or failed onset of female pubertal development (Yatsenko and Rajkovic, 2019). These women have premature ovarian insufficiency (previously referred to as premature ovarian failure). They require hormone replacement therapy with estrogens and usually progestogens to promote pubertal development, support menstrual cycles, and maintain bone health. Typically, these women possess a uterus and can carry a pregnancy using modern reproductive technology, such as in vitro fertilization.
Disorders of androgen excess
The most frequent cause of 46,XX DSD is congenital adrenal hyperplasia, a group of autosomal recessive genetic disorders that affect the adrenal glands (leading to impaired adrenal cortisol biosynthesis and excessive adrenal androgen [C19 steroid] production). A spectrum of phenotypes occurs, reflecting the consequences of the genetic variant for enzyme function, resulting in much clinical heterogeneity. The most common “classic” form of congenital adrenal hyperplasia is 21-hydroxylase deficiency due to deleterious variants in the 21-hydroxylase (CYP21A2) gene. The incidence of the classic (and more severe) form of 21-hydroxylase deficiency (salt-losing and simple virilizing) is approximately 1 in 14,000 to 1 in 18,000 live births worldwide (Claahsen-van der Grinten et al., 2022). The incidence of
the milder (or nonclassic) form is approximately 1 in 200 White American individuals (Hannah-Shmouni et al., 2017). While deficiencies related to 21-hydroxylase account for 90–95 percent of congenital adrenal hyperplasias (Momodu et al., 2023), other enzyme deficiencies that may cause this condition include 11β-hydroxylase deficiency due to CYP11B1 variants, 3β-hydroxysteroid dehydrogenase type 2 deficiency due to HSD3B2 variants, and P450-oxidoreductase deficiency due to POR genetic variants (Miller and Auchus, 2011). Genetic variants in the 17α-hydroxylase/17-20, lyase (CYP17A1) in 46,XX (and 46,XY) individuals are associated with typical female external genital appearance at birth. Because of their inability to produce cortisol, androgens, and estrogens, these individuals may present with hypertension and delayed puberty (Auchus, 2022).
The 21-hydroxylase form of congenital adrenal hyperplasia is typically associated with mineralocorticoid deficiency, which if unrecognized is associated with neonatal deaths and morbidities due to acute adrenal insufficiency (Speiser et al., 2018). Affected 46,XX infants have atypical genitalia at birth. The excessive circulating prenatal androgen concentrations cause virilization of the external genital structures of affected 46,XX fetuses. A small number of 46,XX individuals with 21-hydroxylase deficiency and extensive male external genital development have been assigned male sex at birth (Mazur et al., 2023).
As 21-hydroxylase deficiency is an autosomal recessive disorder, affected 46,XY fetuses have typical male external genitalia at birth (Speiser et al., 2018). Individuals (both 46,XX and 46,XY) with extremely deleterious CYP21A2 genetic variants typically develop acute adrenal insufficiency within the first 2 weeks of life. If these infants are not promptly recognized and treated with appropriate hormone replacement therapy, significant morbidity or mortality may occur. Newborn screening programs exist in all 50 states and many other countries to detect infants with the classic forms of 21-hydroxylase deficiency (Therrell, 2001). Given the possibility of false negative tests in the immediate newborn period, some states have instituted a second screen during the first month of life. However, newborn screening protocols vary widely (Conlon et al., 2023).
Milder (nonclassic) forms of 21-hydroxylase deficiency present in childhood with premature development of pubic hair, phallic (clitoris or penis) enlargement, increased linear growth velocity, and accelerated skeletal maturation. As a result of the nature of the hyperandrogenic symptoms, the mildest form presents predominantly in adolescent and adult women (Claahsen-van der Griten et al., 2022).
Vaginal or uterine atresia
Individuals with uterine/vaginal anomalies may experience pubertal onset with breast development and present for evaluation of primary
amenorrhea and/or hydrometrocolpos (Grant et al., 2023; Porsius et al., 2022). This group of anomalies is labeled as the Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome or Müllerian agenesis, which occurs in approximately 1 in 5,000 46,XX women (Herlin et al., 2020). This syndrome is characterized by incomplete development of the female reproductive tract and may involve the uterus, cervix, and upper vagina. MRKH is subclassified as isolated uterine/vaginal anomalies (Type I) or syndromic (Type II) (Herlin et al., 2020). The syndromic form is associated with other anomalies, such as renal, skeletal, and facial anomalies; the “MURCS” association is a severe form of MRKH characterized by Müllerian duct, renal, and cervicothoracic spine abnormalities. Like people with complete androgen insensitivity syndrome, people with MRKH may seek a uterine transplant to give them the opportunity to bear a biological child (Brännström et al., 2015). Uterine and vaginal agenesis are included in this category. At birth, 46,XX infants with aberrant ovarian development and/or uterine/vaginal anomalies have typical female external genitalia appearance.
46,XX DSD in Males
46,XX males have a male phenotype despite a 46,XX karyotype. There are two different forms of 46,XX DSD in males: SRY (sex-determining region on Y chromosome)–positive or SRY-negative (Wu et al., 2014). Approximately 90 percent of XX males are SRY-positive and have a translocation of the SRY gene to another chromosome, typically the X chromosome. These patients present with micropenis, atypical external genital development, delayed/failed puberty, and/or infertility. The SRY-negative group reflects the consequence of aberrant testicular differentiation. One example of XX sex reversal involves variants in the R-Spondin1 (RSPO1) gene, which is associated with palmoplantar keratoderma, congenital bilateral corneal opacities, nail dystrophy, and hearing impairment (Tomaselli et al., 2008). Other genes associated with SRY-negative XX males include SOX9, SOX3, and FOXL2 (Grinspon and Rey, 2019). One in every 20,000 male births is thought to be 46,XX, and this condition accounts for approximately 2 percent of male infertility cases (Adrião et al., 2020).
Nonhormonal Variations in Sex Traits
In addition to the conditions discussed above and listed in Box 7-1, it is important to recognize disorders associated with atypical genital development that are not caused by hormones or gonadal function. These are primarily anatomic malformation disorders of the genitourinary tract that include caudal regression syndrome, mild epispadias, cloacal exstrophy, omphalocele-exstrophy-imperforate anus-spinal defects complex, and
aphallia. These disorders are usually associated with typical gonadal function congruent with the sex chromosomes. Although these conditions are rare, affected individuals require substantial medical, behavioral health, and surgical treatments to address the structural and functional impairments of the genitourinary, gastrointestinal, and neurologic systems. These conditions typically require lifelong care such that affected individuals may need assistance with activities of daily living either transiently or permanently. These individuals often suffer from urinary and bowel incontinence (Maruf et al., 2020). Urinary incontinence may be treated with a catherizable stoma. Many patients require bowel diversion surgery to achieve bowel continence. Renal anomalies may also coexist. In some instances, the renal anomalies are associated with deteriorating renal function (Bolduc et al., 2002). Skeletal anomalies include scoliosis, hip dysplasia, and clubfeet; affected individuals may require osteotomies. Aberrant development of the uterus and vagina is commonly associated with these malformations. For males, the penis and scrotum are bifid typically with testes palpable within the hemiscrotum. Individuals with these disorders experience numerous challenges, including multiple surgeries, bladder/bowel diversions complicated by foul odor and infections, sexual dysfunction, and embarrassment (Van den Eede et al., 2023). In a group of 63 patients, anxiety and/or depression were noted in 52.4 percent (Haney et al., 2024).
INITIAL MANAGEMENT OF VARIATIONS IN SEX TRAITS
Individuals suspected of having VSTs are often referred to expert and experienced health care professionals. A multidisciplinary team offers the optimal management for such individuals and their families. Members of this team may include pediatric endocrinologists, pediatric urologists/surgeons, geneticists, neonatologists, radiologists, behavioral health specialists, social workers, and pediatric nurse educators (Ahmed et al., 2011; Coleman et al., 2022). The multidisciplinary team provides individualized care in the context of enabling the affected individual to enjoy “the best life possible” (Wilkins et al., 1955; Witchel et al., 2022). Some pediatric hospitals have ongoing family support committees to help with the shared decision making and management of children with VSTs. Figure 7-1 depicts the many types of clinical experts and community supports that may be involved in care for people with VSTs.
Prenatal Diagnosis
VSTs may be suspected prenatally based on fetal ultrasound findings. More recently, noninvasive prenatal screening (NIPS) has increasingly been
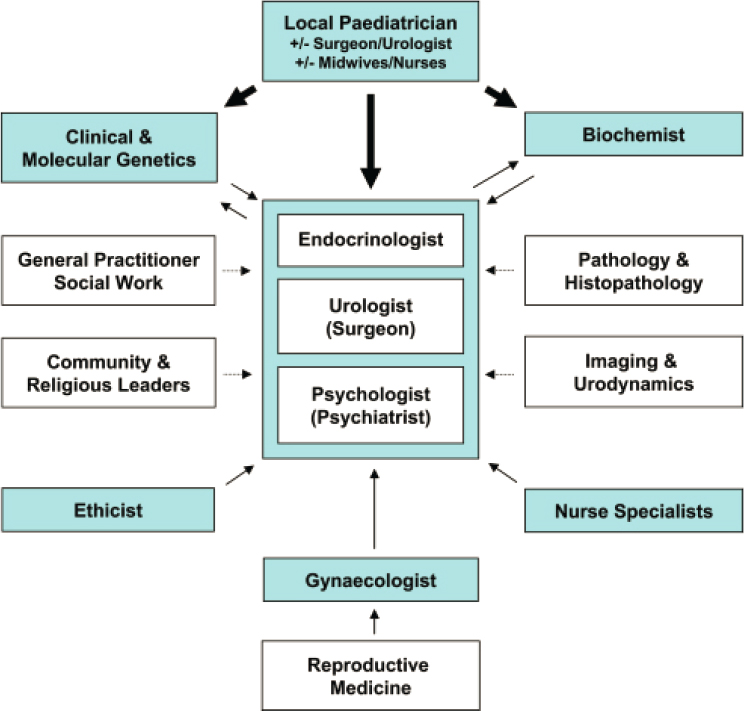
NOTE: Specialists in adolescent medicine are often an important part of the multidisciplinary team. These specialists are included within the local pediatrician box in the schematic.
SOURCE: Brain et al., 2010. CC BY 3.0.
used to determine some VSTs (and other conditions) prenatally (Gregg et al., 2016; Saulnier et al., 2021). This test, which involves a single blood sample obtained from the pregnant person, is used to examine the fetal (placental) cell-free DNA in circulation. NIPS results may differ from those inferred from the ultrasound appearance of the fetal genitalia. When genital ambiguity is evident on the fetal ultrasound exam or discordant results from NIPS/genetic testing are obtained, prenatal consultation with a multidisciplinary team needs to be arranged immediately.
Initial Evaluation by the Multidisciplinary Team
As described above, some people with VSTs are diagnosed prenatally, others at birth, and others later in life (during puberty or adulthood). Regardless of chronologic age at presentation, initial evaluation of VSTs by a multidisciplinary team often includes a comprehensive medical history comprising prenatal, family, and psychosocial histories and information about potential environmental exposures (Brain et al., 2010; Coleman et al., 2022; Cools et al., 2024; Moran and Karkazis, 2012). A thorough physical examination with detailed observation of the external genitalia is performed. Box 7-2 offers examples of common inquiries providers may have during the physical exam.
Next, laboratory and imaging studies, and increasingly genetic studies, are performed. The specific studies performed depend on the patient history and physical examination.
Laboratory Studies
Blood samples are collected to measure hormone concentrations, determine functioning of the adrenal and pituitary glands (adrenocorticotropic hormone stimulation), and/or measure the amount of sex hormones being produced in the body naturally (human chorionic gonadotropin [hCG] stimulation) (Mayo Clinic, 2018). Blood and, occasionally, urine samples are used to determine the karyotype and assess for chromosomal abnormalities. Over time, the methodology for measuring blood and urine hormone concentrations
BOX 7-2
Common Inquiries During Physical Examination to Determine Variations in Sex Traits
- Are the labioscrotal folds fused or not?
- Are gonads palpable?
- Are the external genital structures symmetric?
- What is the appearance of the phallus (clitorophallus)?
- Where is the urethral meatus?
- How many orifices are present on the perineum?
- Are other anomalies present?
- In adolescents and young adults, what clinical features of puberty are present or absent?
SOURCE: Adapted from Ahmed et al., 2011; Lee et al., 2006.
has changed as a result of improved technology. In the past, radioimmunoassays were used to measure hormone concentrations. Given the potential for variable results, these methods are used less commonly today (Braunstein, 2022; Ghazal et al., 2022; Tomlinson et al., 2004). More recently, liquid chromatography–tandem mass spectroscopy is increasingly being used; this method is more accurate and can measure multiple analytes simultaneously (Andrieu et al., 2022; Wudy et al., 2018). However, some hospitals may not have access to liquid chromatography–tandem mass spectroscopy methods. It is important to acknowledge that biotin (a compound commonly used in skin/hair products and vitamins) can interfere with some assays (Samarasinghe et al., 2017).
Imaging Studies
Imaging studies can include ultrasound, magnetic resonance imaging, and computed tomography imaging. The goal of imaging studies is to determine the status of the internal genital structures (e.g., presence or absence of uterus) and to assess for additional anomalies such as renal agenesis (absence of one or both kidneys) (Grinspon et al., 2023). Due to the technical limitations of current imaging modalities, laparoscopic surgery may be necessary to visualize the internal genital structures and, when needed, obtain gonadal biopsies to assess gonadal histology and risk for malignancy (Ahmed et al., 2011; Farrugia et al., 2013).
Genetic Studies
Increasingly, genetic studies are performed as the first-line study, especially in individuals with atypical genitalia and 46,XY karyotypes (Ahmed et al., 2022). Yet despite the increased knowledge and understanding of genetic variants in VSTs, the specific genetic diagnosis remains unknown in many patients (Persani et al., 2022). In addition, not all genetic variants are deleterious or disease-causing. Some genetic variants are characterized as “variants of unknown significance,” which means that available data are insufficient to ascertain the functional significance of a variant (i.e., whether it has any functional consequences for health). Establishing the functional consequences of such variants is often challenging. For this reason, specific standards and guidelines are followed to ascertain the likelihood that a genetic variant is deleterious (Richards et al., 2015).
Sex Assignment at Birth
One of the most difficult aspects of managing patients with VSTs is the sex assignment at birth, also referred to as sex of rearing (i.e., the child’s gender-specific upbringing), which becomes the sex recorded on birth
certificates and medical records. This aspect, and its associated decisions, reflects society’s dogmatic insistence on classifying people by their sex. Through a shared decision-making process between the multidisciplinary care team and the child’s parents or other caregivers, the goal is to select a sex that has the greatest likelihood of matching gender identity in adulthood (Sandberg et al., 2012). However, sex assignment for infants with VSTs is not straightforward, as it is often based on numerous factors, including results of laboratory, imaging, and genetic studies; specific VST diagnosis; phallus length; psychological orientation; the possibility of fertility and sexual functionality; expectation or wishes of the family; and consensus opinion among experienced specialist providers (Campo-Engelstein et al., 2017; Gürbüz et al., 2020; Sandberg et al., 2012). In addition, as discussed previously in this report, an individual’s personal gender identity develops over time.
During initial evaluation of a newborn infant with atypical genitalia, health care providers avoid any language suggesting a “sex assignment” (referring to the baby as “your baby,” rather than “your daughter”/“your son”), and medical documents do not indicate “sex” at this stage. In addition, health care providers explain VSTs to parents or other caregivers with sensitivity to the family’s cultural background (especially related to rituals used to celebrate births), use language at the 5th- to 8th-grade level, and repeat their explanations multiple times (Jackson et al., 2008; Lipstein et al., 2014; Sandberg et al., 2019; Weidler and Peterson, 2019). Early involvement of behavioral/mental health professionals is essential to support the family in dealing with the initial uncertainties regarding sex assignment; the diagnostic procedures; the decision-making process; final diagnosis; and discussions with extended family members, friends, and colleagues. Following these discussions and diagnostic procedures, shared decision-making conversations involving the parents or other caregivers and health care providers take place, ultimately leading to a decision about a sex assignment for the infant. Although some families elect to defer decisions regarding sex of rearing, most families are more comfortable with a binary determination.
Gender Identity
Typically, gender identity follows sex assignment for individuals with VSTs (Bakula et al., 2017; Callens et al., 2016). As defined in Chapter 2, “gender identity” refers to the term used by individuals to label themselves and their internal sense of self. People with VSTs, like anyone else, can have a gender identity that differs from their sex recorded at birth. The proportion of gender identity concerns among people with VSTs is greater than the proportion of TGD people in the general population (Dessens et al., 2005; Furtado et al., 2012; Herman et al., 2022; Hines, 2020; Meyer-Bahlburg et al., 1996; Zucker et al., 1996).
A 2021 systematic review and meta-analysis analyzed the prevalence of transgender or gender diverse (TGD) identity in adolescents and adults with VSTs, finding an overall prevalence of 15 percent (95% CI 13–17 percent), but variability in prevalence of TGD identity among different VST categories (Babu and Shah, 2021). For example, Babu and Shah found that the prevalence of gender dysphoria was 53 percent among populations with 5-alpha reductase deficiency (SRD5A2) who were reared as female, but only 1.7 percent among individuals with complete androgen insensitivity syndrome (all individuals identified in the review with complete androgen insensitivity syndrome were raised as female). This finding mirrors other studies that show TGD identity is rare among individuals with complete androgen insensitivity syndrome (T’Sjoen et al., 2011). However, Babu and Shah found TGD identity to be much higher in populations with partial androgen insensitivity syndrome, with gender dysphoria more common among male-raised individuals compared to female-raised individuals (25 vs. 12 percent). Sex assignment at birth is also an important factor in TGD identity for individuals with congenital adrenal hyperplasia; Babu and Shah found 4 percent gender dysphoria among individuals with congenital adrenal hyperplasia who were reared female, but 15 percent gender dysphoria among those reared male. de Jesus et al. (2019) confirm gender dysphoria is more common in male-raised individuals with congenital adrenal hyperplasia, compared to those raised female.
However, the committee notes that methodological flaws in these studies may obscure what researchers know about gender preferences in VST populations, including the use of invalid and binary measures of gender identity and collection of data when it was much rarer to assert a gender diverse or transgender identity (Pasterski et al., 2015).
As in people without VSTs, the development of the secondary sex characteristics typical of puberty may prompt discussion of gender identity in people with VSTs. Certain VSTs may cause pubertal changes that impact gender identity. For example, some 46,XY infants with aberrant testicular development are undervirilized, and they may be assigned female sex at birth (Thigpen et al., 1992). At puberty, these individuals experience phallic and testicular enlargement and may subsequently self-reassign from female to male (Costa et al., 2012; Maimoun et al., 2011). This specific autosomal recessive disorder, associated with SRD5A2 genetic variants, is more prevalent in specific populations, such as that of the Dominican Republic, where these individuals have been labeled as “guevedoces” (which translates to “penis at 12”) (Marks, 2004).
Upon VST diagnoses, reflection on gender identity may be included in conversations with patients and their care team as these patients may be more likely to question their gender because of their atypical external
Panelist Perspective
“And we make these kind of generalizations based on specific conditions that people have or specific variations that those people have. We also tend to make some broad brushstrokes based on someone’s chromosomes or the presence of certain hormones or you know, specific hormonal levels. But we’re often proven wrong. So, actually, in a clinic setting, although somebody like me who has complete androgen insensitivity syndrome we may typically identify as female most of the time. But then we’ll have a patient, [or] will have a kiddo who comes in, [who] over time starts to identify as nonbinary or start to identify as male. And that would be contrary to what the medical data or a lot of the past research data would tend to tell us or tend to indicate. So, I would just want to caution everyone not to make assumptions about somebody’s gender identity or the way they choose to express themselves, based on their specific traits or based on specific test result[s].”
—Statement from patient–provider panel,
presented to the committee on November 30, 2023.
genitalia, discordant karyotype, fertility status, romantic attractions, or medical interventions (e.g., hormone treatment and/or surgery) (Granero-Molina et al., 2023; Kreukels et al., 2018). Gender identity may be broached during discussions about initiating exogenous sex hormone treatment to induce puberty. In addition, some individuals may seek a gender assessment from a mental health provider, often initiated as a referral from an endocrinologist or medical provider.
The gender-related needs of people with VSTs may be complicated even if a self-initiated gender transition is not desired. For instance, girls with Turner syndrome may feel that they are not true females because of their body differences, need for exogenous estrogen, and likely infertility (Granero-Molina et al., 2023). Others may be concerned about having body parts that they perceive as inconsistent with their affirmed gender. These feelings may lead to poor self-esteem, shame, and confusion. Such concerns are important when considering patients’ psychosocial or mental health needs (Kreukels et al., 2018).
Ongoing Shared Decision Making and Access to Providers and Services
In addition to initial evaluation and decisions about sex assignment, an important function of multidisciplinary care teams is assisting patients
and parents or other caregivers with difficult choices about immediate and future care and treatment (including hormone treatment, surgery, and mental health care, as described in detail below). Health care management recommendations and choices vary even for individuals with similar conditions. Recognizing that health care for VSTs is highly variable and patient specific, coordination of care with patients and their parents or other caregivers is essential among the multidisciplinary providers (Sandberg et al., 2019).
Multidisciplinary care for people with VSTs is clearly beneficial for patients and their parents or other caregivers. Yet access to multidisciplinary care can be challenging because such expertise is limited primarily to large urban areas in the United States. Families may need to travel for many hours to meet with specialists and the multidisciplinary team. Expertise related to specific aspects of VST care may be unavailable in smaller urban and rural areas. In addition, health care providers in rural locations may lack the knowledge and expertise to offer initial VST management because of the rarity of these conditions.
Older persons with VSTs may not have had access to a multidisciplinary care arrangement in their youth and may have lacked the options in care and treatment that exist today. As discussed below, in the past, genital reconstructive surgery was performed during infancy and childhood, reflecting the prevailing hypothesis that genital anatomy needed to be consistent with sex assignment. Due to past recommendations for nondisclosure, some individuals may lack information regarding prior medical or surgical management. For these individuals, ongoing care involves decision making around future medical care and treatment, with particular focus on psychosocial well-being and follow-up care based on earlier surgeries (Berry and Monro, 2022).
HORMONE TREATMENT FOR PEOPLE WITH VARIATIONS IN SEX TRAITS
Hormone treatments may be helpful for some people with VSTs in the context of their gender identity, age, and unique medical circumstances. This section describes appropriate hormone treatment for a number of VST conditions. In addition to the hormone therapies described here, people with VSTs may also access hormone therapy as part of gender-affirming care (described in Chapter 5).
Congenital Adrenal Hyperplasia
Upon confirmation of diagnosis, individuals with congenital adrenal hyperplasia are treated with glucocorticoid replacement therapy. Typically,
hydrocortisone, administered three to four times daily, is used—although other regimens may be appropriate for individual patients (Allolio, 2015; Dineen et al., 2019; Ng et al., 2020). Individuals with the “classic” salt-losing form of congenital adrenal hyperplasia are also treated with fludrocortisone, a synthetic mineralocorticoid. All individuals prescribed daily hydrocortisone treatment must be treated with extra exogenous glucocorticoid for stressful situations such as fever, acute gastroenteritis, and severe physical trauma, as well as during surgery. Initially, parents or other caregivers are responsible for providing this treatment, with eventual training for adolescents and adults. Stress doses can be administered orally if tolerated. If oral medication is not tolerated, parenteral hydrocortisone needs to be administered promptly. Parents or other caregivers and older affected individuals need to have parenteral hydrocortisone readily available and know how and when to administer this medication for acute situations. Individuals requiring hydrocortisone stress dosing need to wear medical alert ID badges (Ahmet et al., 2023; Puar et al., 2016).
Hormone Therapy for Small Penis
During infancy, some boys with a small penis are treated with small doses of testosterone for 3–6 months to promote elongation of the penis (Stancampiano et al., 2022). In some instances, hCG, which is very similar to luteinizing hormone, is administered for an hCG stimulation test to assess testicular testosterone production. Specific practices regarding the number of injections and dosing vary among physicians.
Growth Hormone Therapy
During childhood, sex steroid hormone replacement therapy is unnecessary because sex steroid levels are normally low. For individuals with short stature associated with Turner syndrome, 45,X/46,XY mosaicism, or severe intrauterine growth retardation, growth hormone therapy is beneficial to increase linear growth velocity and final adult height (Bertelloni et al., 2015; Hwang, 2014; Ranke, 1995; Ross et al., 1986; Urban et al., 1979).
Delayed/Absent Puberty
For individuals unable to experience endogenous pubertal development because of hormone deficiencies, sex steroid treatment is used to initiate secondary sex characteristics and promote bone health (De Luca et al., 2001; Klein et al., 2017; Saggese et al., 1997; Villanueva and Argente, 2014). Causes of delayed or absent puberty include hypogonadotropic
hypogonadism, hypothalamic and pituitary tumors, premature ovarian insufficiency, and testicular failure (Howard and Dunkel, 2018; Klein et al., 2017; Sullivan et al., 2016). Some chronic disorders, such as celiac disease, cystic fibrosis, and sickle cell anemia, are commonly associated with delayed puberty (Johannesson et al., 1997; Rhodes et al., 2009; Saari et al., 2015).
Where appropriate, feminizing hormone treatment (small doses of estrogen) is initiated around age 10–14; the estrogen dose is increased gradually (Palmert and Dunkel, 2012). Estrogen is preferably administered using a transdermal estradiol patch; oral and parenteral estrogen are available as alternatives. Approximately 1–2 years following initiation of estrogen treatment, cyclic progestin therapy is initiated when a uterus is present to experience cyclic withdrawal bleeding. Combined estrogen/progestin therapy is important to decrease the risk for endometrial cancer later in life. In addition, experiencing regular menses is important to some patients. Estrogen also promotes uterine growth, which is essential for future pregnancies (when possible).
Where appropriate, masculinizing hormone treatment (small doses of testosterone) is initiated around age 12–15; the testosterone dose is increased gradually (Palmert and Dunkel, 2012). Testosterone is typically administered by intramuscular or subcutaneous injection. Dose titration to initiate puberty is nearly impossible for testosterone gels; in addition, other family members may be accidentally exposed to the testosterone gel. Once an adult testosterone replacement dose has been achieved, testosterone can be administered by intramuscular or subcutaneous injection or transdermal gel. In the future, oral testosterone undecanoate may prove to be beneficial in people with ongoing testosterone deficiency.
Reproduction/Fertility
Gonadotropin deficiencies in patients with congenital or acquired hypogonadotropic hypogonadism result in delayed pubertal development and inadequate stimulation of the gonads to secrete sex steroids and mature gametes (Alexander et al., 2024). Similarly, individuals with disorders affecting steroidogenesis may have hormone imbalances that affect pubertal development and ovulation/spermatogenesis (Sengupta et al., 2021). Depending on the specific diagnosis and endogenous hormone secretion, sex steroid hormone replacement therapy may be needed to promote the development of secondary sex features and maintain bone health.
Gonadal differentiation and pubertal development are abnormal in most individuals with chromosomal anomalies such as 45,X monosomy;
45,X/46,XY; 47,XXY; and other variants. Secondary sex development is delayed/absent because of the deficient secretion of gonadal sex steroid. The aberrant gonadal environment impairs maturation of oogonia and spermatogonia, resulting in subfertility/infertility. Individuals with nonpalpable gonads and Y chromosomal material have an increased risk for gonadal neoplasia. Such individuals typically undergo gonadectomy (Lucas-Herald et al., 2021).
Some individuals, such as those with congenital adrenal hyperplasia, are generally fertile; both women and men may require intensive glucocorticoid hormone replacement therapy to enable normal hypothalamic-pituitary-gonadal axis function and gametogenesis. For some, such as those with Turner syndrome and Klinefelter syndrome, infertility is typical. Depending on the specific disorder, fertility preservation may be possible (Rodriguez-Wallberg et al., 2023).
SURGICAL INTERVENTIONS
Currently, decisions regarding surgical interventions for people with VSTs are complex and involve shared decision making among the patient, parents or other caregivers, and the interdisciplinary health care team. Over the past few decades, philosophies regarding surgical interventions have been discussed and modified. It has been increasingly recognized that even minor operations may have undesirable outcomes. Most importantly, the inability to obtain informed consent from a minor child for a surgical intervention has been acknowledged. Indications for surgery generally focus on functional outcomes rather than aesthetic (Lee et al., 2006). In some instances, for example, early surgical intervention is performed because of an increased risk for urinary tract infections (Ding et al., 2023). Typically, surgery is appropriate for 46,XY individuals with hypospadias to enable standing to urinate and eventually having children (Halaseh et al., 2022). In addition, as described in further detail below, surgery is important for VSTs that cause genitourinary malformation.
While it has been believed that surgery performed for cosmetic reasons in infancy relieves parental stress and improves attachment between the child and parents, this approach lacks consistent evidence (Crawford et al., 2009; Dayner et al., 2004; Lee et al., 2006). Accordingly, many centers have moved away from surgery in early childhood when function is not impaired, and medical organizations oppose medically unnecessary genital surgeries (AAFP, 2024; Children’s Hospital of Chicago, 2021). Historically, however, practices were very different, and older medical records may refer to surgeries performed that were based on best practices
at the time, which, with modern understanding, may no longer be considered appropriate.6
In 2016, the Global DSD Update Consortium released a consensus statement on the approach to and care of individuals with VSTs (Lee et al., 2016). These guidelines reconfirm an obligation for individualized care given that evidence-based data regarding indications, timing, and need for surgery remain uncertain. The authors describe four VST procedures: (1) surgery of the genital tubercle (clitoroplasty or reconstruction), (2) surgery of the Müllerian structures, (3) surgery of the gonads (orchiopexy, removal, biopsy, or preservation), and (4) perineoplasty. When considering each procedure, patient goals, possible complications, and long-term outcomes need to be considered. Despite support for these guidelines by the experts, the levels of evidence for the recommendations were low, and the experts did not reach consensus regarding indication, timing, procedure, and evaluation of outcomes. However, consensus was achieved regarding the following points: (1) health care should be provided by centers of expertise with multidisciplinary care; (2) providers should take a conservative approach to gonadal surgery in patients with complete androgen insensitivity; (3) providers should avoid vaginal dilatation in childhood; (4) asymptomatic Müllerian remnants can remain intact during childhood and removed later if needed; (5) it is appropriate to remove biopsy-confirmed streak gonads among individuals with Y chromosomal material; and (6) in discussions of sex assignment, patients with 46,XY cloacal exstrophy should be raised as males despite anomalous anatomy.
Genitourinary Malformation Syndromes
As noted above, genitourinary malformation syndromes are often included with the differential diagnosis of atypical external genitalia. This category encompasses cloacal exstrophy, bladder exstrophy, and persistent cloaca (also known as urorectal septum malformation sequence). Cloacal
___________________
6 In the 1950s, the American pediatric endocrinology community promoted the belief that a child’s lived experiences as a boy or a girl, established the child’s gender role and erotic orientation and that, at least in the United States, the birth of a child with atypical external genitalia was a social emergency. Parents were advised to share their child’s situation only with family members. These beliefs led to the practice of assigning female sex of rearing to 46,XY infants with an undersized phallus, aphallia, or female-appearing external genital structures. In other words, nurture took precedence over nature in establishing gender identity. The sole exception was girls with congenital adrenal hyperplasia, who are known to have normal female internal genitalia; these virilized girls underwent surgical procedures to make the external genitalia appear more female. Over time, based on lived experiences, challenges arose regarding these hypotheses and the need for early surgical management. Nevertheless, older medical records may need to be reviewed to ascertain any details regarding prior surgeries. Given the past practices of concealment with respect to atypical external genital development and previous genital surgery, individual patients may not be fully aware of past surgical procedures performed during their early childhood.
exstrophy—where a portion of the large intestine lies outside the body—may be part of a more extensive malformation syndrome that includes omphalocele, spinal defects, and imperforate anus (Neel and Tarabay, 2018). It is also commonly associated with other defects of the genitourinary system, such as hydronephrosis/hydroureter, renal agenesis, cystic dysplasia of the kidneys, horseshoe kidney, and duplicated urinary collecting system (Keppler-Noreuil et al., 2017). Bladder exstrophy refers to a defect such that the bladder forms outside the body. Bladder exstrophy typically involves the digestive and reproductive systems as well as the urinary tract; it is often considered to be a milder form of cloacal exstrophy. Both appear to represent different manifestations of a primary developmental field defect in the fetus (Martínez-Frías et al., 2001). Although these are serious conditions and may require a series of operations, the long-term outcome is good for many children with appropriate surgical repair and follow-up care.
Sexual Function
For individuals with atypical external genitalia, sexual intimacy may be challenging as a result of the specific details of an individual’s genital anatomy. In addition, individuals may differ in their definitions and practices for sexual intimacy.
Women with complete androgen insensitivity syndrome, some women with congenital adrenal hyperplasia, women with vaginal atresia, and women with MRKH syndrome may have vaginas inadequate for penile-vaginal intercourse. Vaginal dilatation may be helpful for individuals who desire penile–vaginal intercourse as it may help maintain the dimensions of the vaginal canal (ACOG, 2018; Callens et al., 2014). For some, surgery to construct or lengthen the vaginal canal may be the appropriate intervention. Women with altered anatomy may experience uncomfortable and awkward sexual experiences, and self-esteem and quality of life may be poor (Beisert et al., 2022; Weijenborg et al., 2019). Pelvic floor physical therapy may be beneficial to help alleviate pain and/or assist with psychosocial considerations.
Men with hypospadias, “micropenis,” and other genital malformations may experience difficulty with erection, ejaculation, and orgasm (van der Zwan et al., 2013). Reconstructive surgery may be required to facilitate urinary and/or sexual function.
Sexual discomfort and dysfunction are commonly reported in people with VSTs (Kerckhof et al., 2019; Kohler et al., 2012). While some suggest that complications of surgery are the main factor contributing to dysfunction, this remains uncertain (Crouch et al., 2008; Van de Grift et al., 2022). In a large study of masculinizing surgery, sexual problems and satisfaction with sex life were similarly prevalent in individuals who did and did not undergo surgery (Van de Grift et al., 2022).
Surgical Complications
Complications following surgery for VSTs can be divided into the categories of early and late. Early complications are often related to wound healing and infection, whereas stenosis, stricture, or fistula, as well as recurrent urinary infections, can be seen long term. The need for a second and/or revision surgery is not uncommon. A lack of consistent terminology and the wide range of procedures performed for VSTs make the risk of early complications difficult to quantify.
Also challenging to measure are the long-term outcomes following surgical intervention for VSTs. Outcomes can be classified as both functional and aesthetic (Creighton et al., 2001; Crouch et al., 2008). Functional outcomes, such as voiding and/or sexual dysfunction, are likely related to underlying anatomy, as well as the complexity of the procedures. Aesthetic outcomes can be measured with patient self-reported satisfaction questionnaires. These evaluations are limited by poor response rates, however, and may be biased toward favorable or unfavorable responses (Van de Grift, 2022). Interestingly, in the case of hypospadias repair (surgery to address a problem in the opening of the penis that is present at birth), one study found that adolescents who did not recall the surgery (i.e., had the surgery before 18 months of age) were more satisfied with their overall body appearance compared with those who remembered the surgery (van der Horst and Wall, 2017). Data are inconsistent as to whether hypospadias repair later in life is associated with more complications (van der Horst and Wall, 2017). The multidisciplinary care team needs to engage patients and their families in discussions about possible outcomes, long-term complications of surgery, and quality of life.
MENTAL HEALTH INTERVENTIONS
People with VSTs may experience wide-ranging impacts on their mental and behavioral health and on their social lives. This section discusses important mental health–related considerations for patients with VSTs, relevant risk factors for these patients, mental health considerations for their parents and caregivers, and the role of mental health providers in care for people with VSTs.
Mental Health–Related Considerations for Patients
People with VSTs may have limited access to thoughtful, trained, and knowledgeable health care providers who can support their physical and mental health care needs. This can result in undesirable clinical and psychosocial outcomes. Some patients with VSTs describe stressful or even
traumatic medical practices, including (1) overly frequent and uncomfortable medical/genital exams; (2) lack of privacy due to the presence of multiple providers and trainees in the room; (3) patient marginalization in medical and surgical decision making (often associated with early surgeries, but occurring for adolescents as well); (4) lack of psychosocial support; and (5) stigmatizing communications with health care providers and staff (Haghighat et al., 2023; Thyen et al., 2014). One study found that while satisfaction with health care services was lower for all people with VSTs compared with other patients with serious chronic care needs, satisfaction was lowest among those with the rarest VST conditions because of the scarcity of knowledgeable specialists (Thyen et al., 2014).
Panelist Perspective
“[Patients] encounter providers using insensitive terminology, and also with providers asking invasive questions with a lack of compassion. We are certainly finding out that for many intersex people it is uncomfortable seeking care. Many of the times as intersex adults we don’t know where to seek care in terms of specialty care.”
—Statement from patient–provider panel,
presented to the committee on November 30, 2023.
Navigating social relationships can also be stressful. From the time of diagnosis, many affected individuals and their parents or other caregivers are aware of their physical and medical differences compared with other children. For example, children with VSTs may need to justify to their peers and teachers their repeated absences from school to attend medical appointments. Children with congenital adrenal hyperplasia must wear medical alert ID badges indicating their special needs and must carry emergency medical supplies with them. These factors can lead to absence from important school-related functions, stigmatization, and exclusion (Claahsen-van der Grinten et al., 2022; Traino et al., 2022). Individuals requiring hormone replacement therapy for pubertal development also experience being different from their peers (Dwyer at al., 2019). In addition, some people with VSTs (e.g., patients with genitourinary malformation) need repeated surgeries to address health concerns. The recovery periods associated with such surgeries disrupt both academic and nonacademic activities, and individuals may experience pain and medically related discomforts (e.g., following bladder or bowel diversion procedures). Other VST conditions—particularly
cloacal exstrophy and/or adrenal insufficiency (congenital adrenal hyperplasia)—cause frequent medical crises creating stress in patients and parents (Fleming et al., 2017).
For almost all individuals with VSTs (and their families, as described below), consideration of how and whether to share information about a VST diagnosis with others can provoke anxiety and fear of stigmatization. Children may find it difficult to explain their condition to classmates or teachers, which may hinder social development (Cools et al., 2018). Feelings of shame and fear of negative reactions can lead to social withdrawal, isolation, and poor quality of life (Mackenzie et al., 2009; Schweizer et al., 2009; van Lisdonk, 2014).
Panelist Perspective
“Mental health problems are enormous among adults with intersex variations. And I think a lot of us have come to think of this as partly an artifact of societal stigma and minority stress, but it can for a lot of people also be an artifact of the way they were treated as children navigating medical trauma, navigating complications from the treatments and surgeries that they had as children.”
—Statement from patient–provider panel,
presented to the committee on November 30, 2023.
The pubertal transition from childhood to adulthood leads to particular challenges for adolescents with VSTs, especially for those who are not diagnosed with a VST until puberty fails to progress as expected (Howard and Quinton, 2024). These young people may be confronted with having to reassess their personal identity, sex, gender, medical needs, future goals, and potential for fertility.
In addition to ongoing concerns regarding sexual and urological function, older adolescents and adults often confront fear and apprehension regarding intimacy (Frank, 2018). For example, people with uterine/vaginal agenesis typically must self-dilate to elongate their vaginal “dimple” to engage in sexual intercourse. People with atypical genital development due to congenital adrenal hyperplasia and those with complete androgen insensitivity syndrome may also need to perform self-dilatation. Many feel embarrassed and challenged by this daily task and feel stressed in determining how to reveal their situation to romantic partners before being physically intimate (Batista et al., 2023; Sutton et al., 2005). In many instances, VST diagnoses are accompanied by infertility (as described above), which
can be devastating for the individual and extended family (Diamond and Watson, 2004; Jones, 2020; Sutton et al., 2005). Some people with VSTs experience considerable barriers to accessing knowledgeable medical and mental health care. For some, past negative encounters with the health care system result in avoidance of needed medical care and support (Haghighat et al., 2023; Thyen et al., 2014).
For some people with VSTs, the details of their diagnosis were not shared with them until many years after diagnosis; some did not learn of the diagnosis until adulthood. These practices can contribute to feelings of betrayal, accompanied by reassessment of identity, future prospects, hopes, and dreams (Berry and Monro, 2022; Moreno-Begines et al., 2022). The practice of delayed diagnosis and “keeping secrets” has largely been abandoned by the pediatric endocrine community.
Panelist Perspective
“I think that if you’re going through multiple surgeries at different medical facilities, it is traumatizing. Also, it promotes the challenges of obtaining medical records as adults because you don’t know where to begin, and oftentimes we are not told about having these surgeries [as children] until we are adults, or even much older, when we see a specialist. And we’re not told that this surgical procedure has happened on me.”
—Statement from patient–provider panel,
presented to the committee on November 30, 2023.
Mental Health–Related Risks
As noted above, some people with VSTs experience mental and behavioral health challenges. In one survey of U.S. adults with VSTs (N = 198), more than half of respondents (53.6 percent) described their mental health as fair/poor, 61.1 percent reported depression, 62.6 percent an anxiety disorder, and 40.9 percent posttraumatic stress disorder (PTSD) (Rosenwohl-Mack et al., 2020). Almost a third (31.8 percent) of respondents reported a previous suicide attempt. These results are consistent with those of other studies that have documented high rates of depression, anxiety, and suicide attempts in people with VSTs (Bohet et al., 2019; D’Alberton et al., 2015; de Vries et al., 2019; Engberg et al., 2017; Falhammar et al., 2018; Rosenwohl-Mack et al., 2020; van Rijn et al., 2014). Other commonly reported clinical diagnoses and symptoms among people with VSTs are isolation, stress, low self-esteem, low sexual quality of life (e.g., low sexual
satisfaction, difficulties searching for partners), and feeling they lack identification with a community group (D’Alberton et al., 2015; Schönbucher et al., 2012; Schweizer et al., 2009). However, these findings are only for those individuals willing to participate in such research. Importantly, some people with VSTs choose not to participate in research and published studies may reflect an ascertainment bias. Additional limitations include small sample sizes and heterogeneous disorders. Further research involving more comprehensive patient populations would help clarify the risk factors and identify prevention strategies.
Neuropsychological disabilities and behavioral dysregulation may be associated with specific VST diagnoses and may be noted even before diagnosis of the VST. Individuals with Turner syndrome have an increased prevalence of difficulties with visual-spatial reasoning, visual-spatial memory, executive functioning, and motor and math skills, as well as ADHD; some also have ASD features (Hutaff-Lee et al., 2019). Figure 7-2 depicts the many neuropsychological risks that may impact people with Turner syndrome.
Individuals with Klinefelter syndrome have high rates of bipolar disorder, ASD, ADHD, and psychotic disorders compared with the general population (Bojesen et al., 2006; Bruining et al., 2009; Cederlof et al., 2014; van Rijn et al., 2014). Upon diagnosis of Turner or Klinefelter syndrome, children can be evaluated, monitored, and engaged in early intervention programs to bolster their skills and attempt to prevent risks related to learning or other neuropsychological disabilities (Chadwick et al., 2014;

SOURCE: Hutaff-Lee et al., 2019.
Hutaff-Lee et al., 2019). However, in the past and even at present, few children have been or are regularly monitored or referred for appropriate neuropsychological treatment.
Mental Health–Related Considerations for Family and Caregivers
Specific health care details for people with VSTs depend on multiple factors, such as age at diagnosis; specific VST diagnosis; cultural considerations; and the understanding, beliefs, and attitudes of parents or other caregivers about gender and sex. Upon diagnosis of a VST, parents or other caregivers may feel weighed down by the seemingly overwhelming demands for medical and surgical decision making on behalf of a child. In addition to their beliefs, the availability of counseling and provider biases may influence these complex decisions. Parents or other caregivers may be counseled about their newborn’s VST when simultaneously being exhausted by trying to bond with their baby and meet the baby’s care needs; indeed, they may meet the definition of having PTSD (Duguid et al., 2007; Pasterski et al., 2014).
Panelist Perspective
“The parents of children with variations of sex traits experience really significant and at times disabling mental health distress. There’s some evidence that you compare populations of parents of children with VST relative to parents of children with cancer. Those populations look very similar from a psychological profile and levels of psychological distress.”
—Statement from patient–provider panel,
presented to the committee on November 30, 2023.
Some diagnoses are associated with a range of medical and/or neurodevelopmental disabilities that require monitoring and family support. Parents or other caregivers may be frustrated by the specialized nature of VSTs, the limited number of knowledgeable providers, and worries about their child’s future. In some instances, families must travel to distant health care centers for accurate diagnosis and longitudinal follow-up. Sorting out how medical or surgical decisions may impact choices regarding the child’s name, sex assignment, urinary function, potential fertility, and later sexual function is arduous. Parents or other caregivers may have different opinions and feelings about priorities related to medical and surgical decision making, which can generate family conflict and distrust.
Parents or other caregivers benefit from ongoing education and psychosocial support. However, resources for such support are often inadequate or unavailable for many families, especially in rural settings (Crissman et al., 2011; Fleming et al., 2017). Barriers include limited number of skilled health care providers and lack of financial resources to cover the costs of support (Ernst et al., 2018; Lampalzer et al., 2021). In addition, some health care providers and parents or other caregivers fail to appreciate the benefits of education and psychosocial support.
Roles of Mental Health Providers in Care for People with Variations in Sex Traits
The psychological care needs of people with VSTs and their parents or other caregivers may be wide ranging; specific needs vary among individuals. Ideally, mental health clinicians work as part of the multidisciplinary care team and can ascertain family strengths and vulnerabilities to provide guidance on a child’s developmental needs (Coyne et al., 2023). Mental health providers can help parents or other caregivers understand their beliefs, priorities, and concerns for their children, as well as potential short- and long-term outcomes, in collaboration with medical and surgical clinicians, and can help families resolve conflicts or differences related to health care decisions. In addition, mental health providers can help families decide whether and how to share information about the child’s diagnosis and how to impart developmentally appropriate information to the child. Concerns about what information to share and with whom may vary among family members, and mental health providers can provide support and guidance to help in assessing and resolving differences. Other important areas of discussion may include screening for mental health and emotional needs of parents or other caregivers so they can best support their child. Finally, mental health providers can support families in problem solving and managing other co-occurring life stressors, such as financial, marital, and work–life balance issues.
Older children, adolescents, and adults benefit from similar support from mental health clinicians in monitoring their psychosocial needs and well-being, family and peer supports, and academic progress. Depending on the individual and specific situations, mental health providers can help with VST-related questions that may emerge about gender and identity, fertility and family building, long- and short-term implications of their VST, and past medical and/or surgical interventions. They can help patients process their feelings about their VSTs, normalize differences, and support patients in verbalizing and coping with VST-related stressors and potential stigmatization. They can ensure that VST patients are screened for mental health risks and suicidality, provide interventions as needed, or recommend other
clinicians for long-term therapeutic care. The goal of mental/behavioral health care is to build patient resilience through the development of coping strategies, to empower patients to participate in age-appropriate activities of daily living, to enable patients to educate other providers about their needs, and to teach patients how to self-advocate. Mental health clinicians can provide support for coping with urological and/or sexual dysfunction and the development of peer and intimate relations in the context of having a VST. For people with VSTs who are struggling with age-appropriate activities of daily living, psychological and/or neuropsychological assessment and intervention may be beneficial.
LONG-TERM HEALTH CONCERNS
Patients with VSTs need support in addressing their health concerns long term. Support is especially needed—and sometimes challenging to acquire—when transitioning from pediatric to adult care and when managing co-occurring conditions.
Transition of Care
Ideally, transition of care from pediatric and adolescent health care providers to adult providers needs to be an actively planned process organized by the multidisciplinary care team, addressing medical, psychosocial, educational, and vocational needs of emerging adults (Lee et al., 2006). During childhood and adolescence, parents or other caregivers typically play an active role in the medical management of their children. During the transition period, the individual’s health care paradigm shifts from active supervision to self-directed management, often concomitant with varying education and work settings. Holistic patient-centered comprehensive care throughout the lifespan and especially during the transition from pediatric to adult care maximizes quality of life and promotes the individual’s independence (Balagamage et al., 2023). During the transition process, relevant topics include fertility; sexuality; pathophysiology; knowledge of daily and emergency medications; medical alert ID; and potential chronic consequences, such as obesity, osteopenia, dysglycemia, cardiovascular disease, and testicular adrenal rest tissue in boys and men (Balagamage et al., 2023; Claahsen-van der Griten et al., 2022; Pofi et al., 2023).
Despite the importance of maintaining care regimens—particularly for people who require hormone therapy for management of VSTs—the escalating propensity for risk-taking behaviors in adolescence may lead to poor adherence to medications, office visits, and other aspects of care. Consequences of poor adherence include virilization among girls identified with classic forms of congenital adrenal hyperplasia, short stature, Cushingoid
features,7 and decreased bone mineral density. Individuals who fail to appropriately adhere to glucocorticoid treatment may experience dehydration, hyponatremia, hyperkalemia, hypotension, hypovolemic cardiovascular compromise, or death (Chrisp et al., 2020; Falhammar et al., 2014).
Patients are often lost to follow-up during the transition process (Zahra et al., 2023). For example, previous studies have shown that many young women with Turner syndrome are “lost in transition,” suggesting that successful passage from pediatric to adult care requires a special focus in patients with Turner syndrome (Culen et al., 2017; Davies, 2010).
Additional research is needed to examine pediatric-to-adult health care transitions for people with VSTs to optimize physical and psychosocial outcomes. In addition, appropriate care—particularly mental health services needed by young adults—may not be accessible outside of larger urban areas in the United States and is typically available only in large academic pediatric centers through multidisciplinary teams (Sandberg et al., 2017). This means that for people with VSTs, access to these services becomes scarcer just as they reach maturity.
Panelist Perspective
“[It’s] pretty like darn near impossible, actually, for adults in most parts of the country to be able to access adult providers who know anything about variations in sex characteristics. You create the situation in which people are experiencing health disparities and are not able to access the care they’re getting and what we see in the community is those health problems become increasingly disabling over time for people.”
—Statement from patient–provider panel,
presented to the committee on November 30, 2023.
Management of Co-Occurring Conditions
Cardiovascular Disease
Individuals with 45,X monosomy or other mosaic karyotypes—typically girls diagnosed with Turner syndrome—may have congenital heart disease.
___________________
7 Cushing syndrome occurs when the body has too much of the hormone cortisol for a long time. This condition can result in people with VSTs who take glucocorticoids, which affect the body the same way as cortisol. Symptoms include a fatty hump between the shoulders, a rounded face, and pink or purple stretch marks on the skin.
Common disorders in these individuals include coarctation of the aorta, bicuspid aortic valve, and partial anomalous pulmonary venous return (Meccanici et al., 2023). Older individuals with Turner syndrome have a high propensity to develop dilatated aortic valves and aortic dissection, the latter being a catastrophic event with a high mortality rate (Thunström et al., 2023). Although congenital heart disease and cardiac surgery are more common in individuals with 45,X monosomy compared with those with other karyotypes, the risk of developing aortic dilatation is similar, necessitating regular cardiovascular surveillance (Birjiniuk et al., 2023). Cardiovascular surveillance necessitates cardiac echograms in infants and young children; adolescents and adults need cardiac magnetic resonance angiography studies.
Individuals with congenital adrenal hyperplasia have an increased risk for cardiometabolic disease. This risk is attributed to excessive glucocorticoid dosing, obesity, insulin resistance, hypertension, and dyslipidemia (Torky et al., 2021).
Hypertension is frequently identified in girls, adolescents, and adults with Turner syndrome. The etiology of hypertension in Turner syndrome is likely multifactorial; contributing factors include congenital heart disease, renal anomalies, impaired vagal tone, and high body mass index (McCarrison et al., 2023). Individuals with 21-hydroxylase deficiency congenital adrenal hyperplasia have a higher risk of developing hypertension compared with the general population. Contributing factors include excessive fludrocortisone dosage and obesity (Espinosa Reyes et al., 2023; Falhammar et al., 2011).
Bone Health
Osteopenia and osteoporosis can occur among people with VSTs. For congenital adrenal hyperplasia and other disorders requiring chronic glucocorticoid replacement therapy, monitoring of bone mass density by dual-energy X-ray absorptiometry is warranted, and treatment with calcium, vitamin D, and bisphosphonate drugs may be indicated. Individuals with congenital adrenal hyperplasia diagnosed prior to the introduction of routine newborn screening have an increased prevalence of both any fractures and fractures associated with osteoporosis (Falhammar et al., 2022).
Individuals with sex steroid hormone deficiencies, such as those associated with Turner syndrome and other VSTs, have an increased risk of osteopenia and osteoporosis. Fracture and osteoporosis pose major health challenges for women with Turner syndrome. Although estrogen hormone replacement therapy improves bone mineral density, this appropriate hormone treatment does not fully mitigate against bone loss (Ikegawa and Hasegawa, 2022).
Neoplasia
Aberrant testicular development, also known as testicular dysgenesis, and aberrant gonadal development in the presence of a Y chromosome is associated with an increased risk for gonadal germ cell tumors. For this reason, gonadectomy is performed. Gonadoblastoma have been described in infants at 3 and 5 months of age (Berklite et al., 2019). Individuals with nonpalpable gonads and Y chromosomal material have an increased risk for gonadal neoplasia. Such individuals typically undergo gonadectomy and consequently are infertile (Lucas-Herald et al., 2021; Mittal et al., 2023).
Neuropsychological and Learning-Related Needs
Girls with Turner syndrome typically have normal intelligence, apart from specific areas such as geometry and spatial relations. Some have developmental delay and autism (Björlin Avdic et al., 2021; Kremen et al., 2023). Phenotype–genotype correlation regarding intellectual abilities is poor. Boys with 47,XXY or 47,XYY karyotypes tend to have learning difficulties, psychosocial issues, and increased risk for autism (Jordan et al., 2023; Tartaglia et al., 2017). Children with congenital adrenal hyperplasia who have experienced multiple episodes of acute adrenal insufficiency with electrolyte abnormalities may have learning difficulties (Berenbaum, 2001).
Autoimmune Disorders
Women with Turner syndrome are at increased risk for autoimmune disorders, especially Hashimoto’s hypothyroidism (Bakalov et al., 2012; Naessén et al., 2024). Men with Klinefelter syndrome (47,XXY) also have an increased risk of autoimmune disorders (Kanakis and Nieschlag, 2018; Seminog et al., 2015).
Metabolic Syndromes
The reported prevalence of obesity among individuals with congenital adrenal hyperplasia varies among populations. Abnormal glucose tolerance, dyslipidemia, increased thromboembolic risk, and increased carotid intima thickness have been described in these patients (Righi et al., 2023).
Other Conditions Included in This Report
Chapters 8 through 12 of this report describe a number of specific conditions investigated by this committee, including respiratory disease (asthma, chronic obstructive pulmonary disease, cystic fibrosis, and others),
childhood growth failure, chronic kidney disease, cancers of the reproductive system, and certain gynecological manifestations of HIV. Overall, few studies have examined these conditions among people with VSTs. However, these chapters present a few findings. Chapter 8 describes some findings about the prevalence of respiratory disease among VST populations—for example, a study found that androgen insensitivity syndrome is strongly associated with increased asthma risk (Gaston et al., 2021). Studies have also found that men with Klinefelter syndrome are more likely to be diagnosed with pulmonary diseases than age-matched male controls (Bojesen et al., 2006), and cystic fibrosis has been linked with male hypogonadism (Yoon et al., 2019). Chapter 9 examines certain VSTs known to interfere with childhood growth, including Turner syndrome. Chapter 10 describes how some people with VSTs, such as people with testosterone deficiency (hypogonadism), may be at elevated risk of chronic kidney disease (Romejko et al., 2022); testosterone deficiency is common in patients receiving dialysis (Carrero and Stenvinkel, 2012; Edey, 2017). In addition, some women with Turner syndrome may develop impaired kidney function over time (Izumita et al., 2020; Ogawa et al., 2021). As described in Chapter 11, the literature evaluating the incidence and experience of cancers of the reproductive organs among people with VSTs is exceptionally limited and insufficient for drawing conclusions. Finally, as described in Chapter 12, the epidemiology of HIV among people with VSTs is unknown.
SUMMARY OF KEY POINTS
People with VSTs are a heterogenous group that includes individuals with genetic variations and malformation syndromes affecting the anatomy of the genitourinary and reproductive systems. The diverse VST diagnoses differ in extent and impact on health and chronic disease. Some people with VSTs experience lifelong and ongoing care needs, whereas others experience sporadic severe health crises (for example, cycling in and out of health crises or experiencing a sudden need for lengthy hospitalization, followed by periods of stability). Some individuals experience mental health conditions and neurodiversity associated with VST-related stressors. Details of health care and treatments are highly variable and patient specific, and currently, multiple gaps exist in the research surrounding health care for the VST patient population, producing an imperfect standardized approach to care. Further research is necessary to gain a deeper understanding of the impacts and implications of VSTs across a range of chronic conditions.
As examined in Part III of this report, access to and timing of services related to care for VSTs—in particular, hormone therapy that may be provided during puberty or later in life—may impact functional assessment for people with VSTs who have chronic care needs. Ideally, care for people with VSTs
involves a multidisciplinary team of providers that communicates well and guides patients through all their care needs, including care for chronic disease. However, many individuals, especially in the past, have encountered substantial barriers to consistent multidisciplinary health care and lacked access to knowledgeable providers. Hence, thorough documentation of care related to past or ongoing VST treatment may not be present in medical records. In addition, medical records that merely list a VST diagnosis may fail to provide sufficient historical information for evaluating health and functional status.
Having sufficiently complete medical records that document care related to management of VSTs is important for disability adjudication for applicants with VSTs. Given that a VST diagnosis is often made during early childhood and adolescence, records from many years ago may be relevant to disability adjudication for these individuals. In addition, although available SSA data do not allow for analysis of physical and mental health conditions (“impairments”) among beneficiaries with VSTs, given the body of evidence presented in this chapter that people with VSTs often experience a high burden of physical and mental health conditions, people with VSTs may be particularly likely to present to SSA with multiple impairments. Because SSA considers the combined effects of all impairments when determining eligibility for benefits (SSA, 2017),8 records related to other mental and physical chronic health conditions experienced by applicants with VSTs are important.
Given that patients with VSTs and their parents or other caregivers often lack access to supportive health care services, SSA may better serve applicants with VSTs by explaining to them that medical records related to VST care and related chronic health conditions may be relevant to their disability application. However, the committee acknowledges that older records related to care for VSTs may be very difficult to obtain, and an undue burden would result by requiring applicants to locate such records.
REFERENCES
AAFP (American Academy of Family Physicians). 2024. Genital surgeries in intersex children. https://www.aafp.org/about/policies/all/genital-surgeries.html (accessed March 12, 2024).
Aaronson, I. A., and A. J. Aaronson. 2010. How should we classify intersex disorders? Journal of Pediatric Urology 6(5):443–446.
Acién, P., and M. Acién. 2020. Disorders of sex development: Classification, review, and impact on fertility. Journal of Clinical Medicine 9(11):3555.
ACOG (American College of Obstetricians and Gynecologists). 2018. Müllerian agenesis: Diagnosis, management, and treatment. https://www.acog.org/clinical/clinical-guidance/committee-opinion/articles/2018/01/mullerian-agenesis-diagnosis-management-and-treatment (accessed March 12, 2024).
Adrião, M., S. Ferreira, R. S. Silva, M. Garcia, S. Dória, C. Costa, C. Castro-Correia, and M. Fontoura. 2020. 46,XX male disorder of sexual development. Clinical Pediatric Endocrinology 29(1):43–45.
___________________
8 20 C.F.R. § 404.1523 (2017).
Ahmed, S. F., J. C. Achermann, W. Arlt, A. H. Balen, G. Conway, Z. L. Edwards, S. Elford, I. A. Hughes, L. Izatt, N. Krone, H. L. Miles, S. O’Toole, L. Perry, C. Sanders, M. Simmonds, A. M. Wallace, A. Watt, and D. Willis. 2011. UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clinical Endocrinology 75(1):12–26.
Ahmed, S. F., M. Alimusina, R. L. Batista, S. Domenice, N. Lisboa Gomes, R. McGowan, S. Patjamontri, and B. B. Mendonca. 2022. The use of genetics for reaching a diagnosis in XY DSD. Sexual Development 16(2-3):207–224.
Ahmet, A., A. Gupta, J. Malcolm, and C. Constantacos. 2023. Approach to the patient: Preventing adrenal crisis through patient and clinician education. Journal of Clinical Endocrinology & Metabolism 108(7):1797–1805.
Alexander, E. C., D. Faruqi, R. Farquhar, A. Unadkat, K. Ng Yin, R. Hoskyns, R. Varughese, and S. R. Howard. 2024. Gonadotropins for pubertal induction in males with hypogonadotropic hypogonadism: Systematic review and meta-analysis. European Journal of Endocrinology 190(1):S1–S11.
Allolio, B. 2015. Extensive expertise in endocrinology: Adrenal crisis. European Journal of Endocrinology 172(3):R115–R124.
Andrieu, T., T. du Toit, B. Vogt, M. D. Mueller, and M. Groessl. 2022. Parallel targeted and non-targeted quantitative analysis of steroids in human serum and peritoneal fluid by liquid chromatography high-resolution mass spectrometry. Analytical and Bioanalytical Chemistry 414(25):7461–7472.
Auchus, R. J. 2022. The uncommon forms of congenital adrenal hyperplasia. Current Opinion in Endocrinology, Diabetes and Obesity 29(3):263–270.
Augoulea, A., G. Zachou, and I. Lambrinoudaki. 2019. Turner syndrome and osteoporosis. Maturitas 130:41–49.
Babu, R., and U. Shaw. 2021. Gender identity disorder (GID) in adolescents and adults with differences of sex development (DSD): A systematic review and meta-analysis. Journal of Pediatric Urology 17(1):39–47.
Bakalov, V. K., L. Gutin, C. M. Cheng, J. Zhou, P. Sheth, K. Shah, S. Arepalli, V. Vanderhoof, L. M. Nelson, and C. A. Bondy. 2012. Autoimmune disorders in women with Turner syndrome and women with karyotypically normal primary ovarian insufficiency. Journal of Autoimmunity 38(4):315–321.
Bakula, D. M., A. J. Mullins, C. M. Sharkey, C. Wolfe-Christensen, L. L. Mullins, and A. B. Wisniewski. 2017. Gender identity outcomes in children with disorders/differences of sex development: Predictive factors. Seminars in Perinatology 41(4):214–217.
Balagamage, C., A. Arshad, Y. S. Elhassan, W. Ben Said, R. E. Krone, H. Gleeson, and J. Idkowiak. 2023. Management aspects of congenital adrenal hyperplasia during adolescence and transition to adult care. Clinical Endocrinology. Advanced online publication. https://doi.org/10.1111/cen.14992.
Barros, B. A., L. R. Oliveira, C. R. C. Surur, A. A. Barros-Filho, A. T. Maciel-Guerra, and G. Guerra-Junior. 2021. Complete androgen insensitivity syndrome and risk of gonadal malignancy: Systematic review. Annals of Pediatric Endocrinology & Metabolism 26(1):19–23.
Batista, R. L., M. Inácio, V. N. Brito, M. H. P. Sircili, M. J. Bag, N. L. Gomes, E. M. F. Costa, S. Domenice, and B. B. Mendonca. 2023. Sexuality and fertility desire in a large cohort of individuals with 46,XY differences in sex development. Clinics (Sao Paulo, Brazil) 78:100185.
Beisert, M. J., A. M. Chodecka, K. Walczyk-Matyja, M. E. Szymaêska-Pytliêska, W. Kędzia, and K. Kapczuk. 2022. Psychological correlates of sexual self-esteem in young women with Mayer-Rokitansky-Küster-Hauser syndrome. Current Issues in Personality Psychology 10(4):333–342.
Berenbaum, S. A. 2001. Cognitive function in congenital adrenal hyperplasia. Endocrinology & Metabolism Clinics of North America 30(1):173–192.
Berglund, A., K. Stochholm, and C. H. Gravholt. 2020. The epidemiology of sex chromosome abnormalities. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 184(2):202–215.
Bergougnoux, A., L. Gaspari, M. Soleirol, N. Servant, S. Soskin, S. Rossignol, K. Wagner-Mahler, J. Bertherat, C. Sultan, N. Kalfa, and F. Paris. 2023. Virilization at puberty in adolescent girls may reveal a 46,XY disorder of sexual development. Endocrine Connections 12(12):e230267.
Berklite, L., S. F. Witchel, S. A. Yatsenko, F. X. Schneck, and M. Reyes-Mugica. 2019. Early bilateral gonadoblastoma associated with 45,X/46,XY mosaicism: The spectrum of undifferentiated gonadal tissue and gonadoblastoma in the first months of life. Pediatric and Developmental Pathology 22(4):380–385.
Berry, A. W., and S. Monro. 2022. Ageing in obscurity: A critical literature review regarding older intersex people. Sexual and Reproductive Health Matters 30(1):e2136027.
Bertelloni, S., G. I. Baroncelli, F. Massart, and B. Toschi. 2015. Growth in boys with 45,X/46,XY mosaicism: Effect of growth hormone treatment on statural growth. Sexual Development 9(4):183–189.
Birjiniuk, A., A. G. Weisman, C. Laternser, J. Camarda, W. J. Brickman, R. Habiby, and S. R. Patel. 2023. Cardiovascular manifestations of Turner syndrome: Phenotypic differences between karyotype subtypes. Pediatric Cardiology 45(7):1407–1414.
Björlin Avdic, H., A. Butwicka, A. Nordenström, C. Almqvist, A. Nordenskjöld, H. Engberg, and L. Frisén. 2021. Neurodevelopmental and psychiatric disorders in females with Turner syndrome: A population-based study. Journal of Neurodevelopmental Disorders 13(1):51.
Blackless, M., A. Charuvastra, A. Derryck, A. Fausto-Sterling, K. Lauzanne, and E. Lee. 2000. How sexually dimorphic are we?: Review and synthesis. American Journal of Human Biology 12(2):151–166.
Bohet, M., R. Besson, R. Jardri, S. Manouvrier, S. Catteau-Jonard, M. Cartigny, E. Aubry, C. Leroy, C. Frochisse, and F. Medjkane. 2019. Mental health status of individuals with sexual development disorders: A review. Journal of Pediatric Urology 15(4):356–366.
Bojesen, A., S. Juul, N. H. Birkebaek, and C. H. Gravholt. 2006. Morbidity in Klinefelter syndrome: A Danish register study based on hospital discharge diagnoses. Journal of Clinical Endocrinology & Metabolism 91(4):1254–1260.
Bolduc, S., G. Capolicchio, J. Upadhyay, D. J. Bagli, A. E. Khoury, and G. A. McLorie. 2002. The fate of the upper urinary tract in exstrophy. Journal of Urology 168(6):2579–2582.
Brain, C. E., S. M. Creighton, I. Mushtaq, P. A. Carmichael, A. Barnicoat, J. W. Honour, V. Larcher, and J. C. Achermann. 2010. Holistic management of DSD. Best Practice & Research Clinical Endocrinology & Metabolism 24(2):335–354.
Brännström, M., L. Johannesson, H. Bokstrom, N. Kvarnstrom, J. Molne, P. Dahm-Kahler, A. Enskog, M. Milenkovic, J. Ekberg, C. Diaz-Garcia, M. Gabel, A. Hanafy, H. Hagberg, M. Olausson, and L. Nilsson. 2015. Livebirth after uterus transplantation. The Lancet 385(9968):607–616.
Braunstein, G. D. 2022. Spurious serum hormone immunoassay results: Causes, recognition, management. touchREVIEWS in Endocrinology 18(2):141–147.
Bruining, H., H. Swaab, M. Kas, and H. van Engeland. 2009. Psychiatric characteristics in a self-selected sample of boys with Klinefelter syndrome. Pediatrics 123(5):e865–e870.
Brunello, F. G., and R. A. Rey. 2022. AMH and AMHR2 involvement in congenital disorders of sex development. Sexual Development 16(2-3):138–146.
Callens, N., G. De Cuypere, P. De Sutter, S. Monstrey, S. Weyers, P. Hoebeke, and M. Cools. 2014. An update on surgical and non-surgical treatments for vaginal hypoplasia. Human Reproduction Update 20(5):775–801.
Callens, N., M. Van Kuyk, J. H. van Kuppenveld, S. L. S. Drop, P. T. Cohen-Kettenis, A. B. Dessens, and the Dutch Study Group on DSD. 2016. Recalled and current gender role behavior, gender identity and sexual orientation in adults with disorders/differences of sex development. Hormones and Behavior 86:8–20.
Campo-Engelstein, L., D. Chen, A. B. Baratz, E. K. Johnson, and C. Finlayson. 2017. The ethics of fertility preservation for pediatric patients with differences (disorders) of sex development. Journal of the Endocrine Society 1(6):638–645.
Carrero, J. J., and P. Stenvinkel. 2012. The vulnerable man: Impact of testosterone deficiency on the uraemic phenotype. Nephrology Dialysis Transplantation 27(11):4030–4041.
Cederlof, M., A. Ohlsson Gotby, H. Larsson, E. Serlachius, M. Boman, N. Langstrom, M. Landen, and P. Lichtenstein. 2014. Klinefelter syndrome and risk of psychosis, autism and ADHD. Journal of Psychiatric Research 48(1):128–130.
Chadwick, P. M., A. Smyth, and L. M. Liao. 2014. Improving self-esteem in women diagnosed with Turner syndrome: Results of a pilot intervention. Journal of Pediatric & Adolescent Gynecology 27(3):129–132.
Chaudhry, S., R. Tadokoro-Cuccaro, S. E. Hannema, C. L. Acerini, and I. A. Hughes. 2017. Frequency of gonadal tumours in complete androgen insensitivity syndrome (CAIS): A retrospective case-series analysis. Journal of Pediatric Urology 13(5):e491–e498.
Chen, Z., P. Li, Y. Lyu, Y. Wang, K. Gao, J. Wang, F. Lan, and F. Chen. 2023. Molecular genetics and general management of androgen insensitivity syndrome. Intractable & Rare Diseases Research 12(2):71–77.
Children’s Hospital of Chicago. 2021. Update on intersex care at Lurie Children’s and our sex development clinic. https://www.luriechildrens.org/en/blog/update-on-intersex-care-at-lurie-childrens-and-our-sex-development-clinic (accessed March 12, 2024).
Chrisp, G. L., D. J. Torpy, A. M. Maguire, M. Quartararo, H. Falhammar, B. R. King, C. F. Munns, S. Hameed, and R. L. Rushworth. 2020. The effect of patient-managed stress dosing on electrolytes and blood pressure in acute illness in children with adrenal insufficiency. Clinical Endocrinology 93(2):97–103.
Claahsen-van der Grinten, H. L., P. W. Speiser, S. F. Ahmed, W. Arlt, R. J. Auchus, H. Falhammar, C. E. Fluck, L. Guasti, A. Huebner, B. B. M. Kortmann, N. Krone, D. P. Merke, W. L. Miller, A. Nordenstrom, N. Reisch, D. E. Sandberg, N. Stikkelbroeck, P. Touraine, A. Utari, S. A. Wudy, and P. C. White. 2022. Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocrine Reviews 43(1):91–159.
Coleman, E., A. E. Radix, W. P. Bouman, G. R. Brown, A. L. C. de Vries, M. B. Deutsch, R. Ettner, L. Fraser, M. Goodman, J. Green, A. B. Hancock, T. W. Johnson, D. H. Karasic, G. A. Knudson, S. F. Leibowitz, H. F. L. Meyer-Bahlburg, S. J. Monstrey, J. Motmans, L. Nahata, T. O. Nieder, S. L. Reisner, C. Richards, L. S. Schechter, V. Tangpricha, A. C. Tishelman, M. A. A. Van Trotsenburg, S. Winter, K. Ducheny, N. J. Adams, T. M. Adrián, L. R. Allen, D. Azul, H. Bagga, K. Başar, D. S. Bathory, J. J. Belinky, D. R. Berg, J. U. Berli, R. O. Bluebond-Langner, M. B. Bouman, M. L. Bowers, P. J. Brassard, J. Byrne, L. Capitán, C. J. Cargill, J. M. Carswell, S. C. Chang, G. Chelvakumar, T. Corneil, K. B. Dalke, G. De Cuypere, E. de Vries, M. Den Heijer, A. H. Devor, C. Dhejne, A. D’Marco, E. K. Edmiston, L. Edwards-Leeper, R. Ehrbar, D. Ehrensaft, J. Eisfeld, E. Elaut, L. Erickson-Schroth, J. L. Feldman, A. D. Fisher, M. M. Garcia, L. Gijs, S. E. Green, B. P. Hall, T. L. D. Hardy, M. S. Irwig, L. A. Jacobs, A. C. Janssen, K. Johnson, D. T. Klink, B. P. C. Kreukels, L. E. Kuper, E. J. Kvach, M. A. Malouf, R. Massey, T. Mazur, C. McLachlan, S. D. Morrison, S. W. Mosser, P. M. Neira, U. Nygren, J. M. Oates, J. Obedin-Maliver, G. Pagkalos, J. Patton, N. Phanuphak, K. Rachlin, T. Reed, G. N. Rider, J. Ristori, S. Robbins-Cherry, S. A. Roberts, K. A. Rodriguez-Wallberg, S. M. Rosenthal, K. Sabir, J. D. Safer, A. I. Scheim, L. J. Seal, T. J. Sehoole, K. Spencer, C. St. Amand, T. D. Steensma, J. F. Strang, G. B. Taylor, K. Tilleman, G. G. T’Sjoen, L. N. Vala, N. M. Van Mello, J. F. Veale, J. A. Vencill, B. Vincent, L. M. Wesp, M. A. West, and J. Arcelus. 2022. Standards of care for the health of transgender and gender diverse people, version 8. International Journal of Transgender Health 23(Suppl 1):S1–S259.
Conlon, T. A., C. P. Hawkes, J. Brady, J. G. Loeber, and N. Murphy. 2023. International newborn screening practices for the early detection of congenital adrenal hyperplasia. Hormone Research in Paediatrics 97(2):113–125.
Cools, M., A. Nordenström, R. Robeva, J. Hall, P. Westerveld, C. Flück, B. Köhler, M. Berra, A. Springer, K. Schweizer, V. Pasterski, and COST Action BM 1303 Working Group 1. 2018. Caring for individuals with a difference of sex development (DSD): A consensus statement. Nature Reviews Endocrinology 14(7):415–429.
Cools, M., E. Y. Cheng, J. Hall, J. Alderson, A. M. Amies Oelschlager, A. H. Balen, Y. M. Chan, M. E. Geffner, C. H. Gravholt, T. Guran, P. Hoebeke, P. Lee, E. Magritte, D. Matos, K. McElreavey, H. F. L. Meyer-Bahlburg, R. C. Rink, A. Springer, K. M. Szymanski, E. Vilain, J. Williams, K. P. Wolffenbuttel, D. E. Sandberg, and R. Subramaniam. 2024. Multistakeholder opinion statement on the care of individuals born with differences of sex development: Common ground and opportunities for improvement. Hormone Research in Paediatrics 1–17.
Costa, E. M., S. Domenice, M. H. Sircili, M. Inacio, and B. B. Mendonca. 2012. DSD due to 5alpha-reductase 2 deficiency: From diagnosis to long term outcome. Seminars in Reproductive Medicine 30(5):427–431.
Cox, K., J. Bryce, J. Jiang, M. Rodie, R. Sinnott, M. Alkhawari, W. Arlt, L. Audi, A. Balsamo, S. Bertelloni, M. Cools, F. Darendeliler, S. Drop, M. Ellaithi, T. Guran, O. Hiort, P. M. Holterhus, I. Hughes, N. Krone, L. Lisa, Y. Morel, O. Soder, P. Wieacker, and S. F. Ahmed. 2014. Novel associations in disorders of sex development: Findings from the I-DSD registry. Journal of Clinical Endocrinology & Metabolism 99(2):E348–E355.
Coyne C. A., B. T. Yuodsnukis, and D. Chen. 2023. Gender dysphoria: Optimizing healthcare for transgender and gender diverse youth with a multidisciplinary approach. Neuropsy-chiatric Disease and Treatment 19:479–493.
Crawford, J. M., G. Warne, S. Grover, B. R. Southwell, and J. M. Hutson. 2009. Results from a pediatric surgical centre justify early intervention in disorders of sex development. Journal of Pediatric Surgery 44(2):413–416.
Creighton, S. M., C. L. Minto, and S. J. Steele. 2001. Objective cosmetic and anatomical outcomes at adolescence of feminising surgery for ambiguous genitalia done in childhood. The Lancet 358(9276):124–125.
Crissman, H. P., L. Warner, M. Gardner, M. Carr, A. Schast, A. L. Quittner, B. Kogan, and D. E. Sandberg. 2011. Children with disorders of sex development: A qualitative study of early parental experience. International Journal of Pediatric Endocrinology 2011(1):10.
Crouch, N. S., L. M. Liao, C. R. Woodhouse, G. S. Conway, and S. M. Creighton. 2008. Sexual function and genital sensitivity following feminizing genitoplasty for congenital adrenal hyperplasia. Journal of Urology 179(2):634–638.
Culen, C., D. A. Ertl, K. Schubert, L. Bartha-Doering, and G. Haeusler. 2017. Care of girls and women with Turner syndrome: Beyond growth and hormones. Endocrine Connections 6(4):R39–R51.
Da Aw, L., M. M. Zain, S. C. Esteves, and P. Humaidan. 2016. Persistent Mullerian duct syndrome: A rare entity with a rare presentation in need of multidisciplinary management. International Brazilian Journal of Urology 42(6):1237–1243.
D’Alberton, F., M. T. Assante, M. Foresti, A. Balsamo, S. Bertelloni, E. Dati, L. Nardi, M. L. Bacchi, and L. Mazzanti. 2015. Quality of life and psychological adjustment of women living with 46,XY differences of sex development. Journal of Sexual Medicine 12(6):1440–1449.
Davies, J. H., E. J. Knight, A. Savage, J. Brown, and P. S. Malone. 2011. Evaluation of terminology used to describe disorders of sex development. Journal of Pediatric Urology 7(4):412–415.
Davies, M. C. 2010. Lost in transition: The needs of adolescents with Turner syndrome. British Journal of Obstetrics and Gynaecology 117(2):134–136.
Davis, S. M., L. Bloy, T. P. L., Roberts, K. Kowal, A. Alston, A. Tahsin, A. Truxon, and J. L., Ross. 2020. Testicular function in boys with 47,XYY and relationship to phenotype. American Journal of Medical Genetics: Part C, Seminars in Medical Genetics 184(2):371–385.
Davis, S. M., C. Teerlink, J. A. Lynch, B. R. Gorman, M. Pagadala, A. Liu, M. S. Panizzon, V. C. Merritt, G. Genovese, S. Pyarajan, J. L. Ross, and R. L. Hauger. 2023. Prevalence, morbidity, and mortality of 1,609 men with sex chromosome aneuploidy: Results from the diverse Million Veteran Program cohort. medRxiv (Cold Spring Harbor Laboratory). Advanced online publication. https://doi.org/10.1101/2023.07.15.23292710.
Dayner, J. E., P. A. Lee, and C. P. Houk. 2004. Medical treatment of intersex: Parental perspectives. Journal of Urology 172(4 Pt 2):1762–1765.
de Jesus, L. E., E. C. Costa, and S. Dekermacher. 2019. Gender dysphoria and XX congenital adrenal hyperplasia: How frequent is it? Is male-sex rearing a good idea? Journal of Pediatric Surgery 54(11):2421–2427.
De Luca, F., J. Argente, L. Cavallo, E. Crowne, H. A. Delemarre-Van de Waal, C. De Sanctis, S. Di Maio, E. Norjavaara, W. Oostdijk, F. Severi, G. Tonini, G. Trifiro, P. G. Voorhoeve, F. Wu, and the International Workshop on Management of Puberty for Optimum Auxological Results. 2001. Management of puberty in constitutional delay of growth and puberty. Journal of Pediatric Endocrinology & Metabolism 2(Suppl l4):953–957.
De Sanctis, V., and D. Khater. 2019. Autoimmune diseases in Turner syndrome: An overview. Acta Biomedica 90(3):341–344.
de Vries, A. L. C., R. Roehle, L. Marshall, L. Frisén, T. C. van de Grift, B. P. C. Kreukels, C. Bouvattier, B. Kohler, U. Thyen, A. Nordenström, M. Rapp, and P. T. Cohen-Kettenis. 2019. Mental health of a large group of adults with disorders of sex development in six eastern European countries. Psychosomatic Medicine 81(7):629–640.
Dessens, A. B., F. M. Slijper, and S. L. Drop. 2005. Gender dysphoria and gender change in chromosomal females with congenital adrenal hyperplasia. Archives of Sexual Behavior 34(4):389–397.
Diamond, M., and L. A. Watson. 2004. Androgen insensitivity syndrome and Klinefelter’s syndrome: Sex and gender considerations. Child and Adolescent Psychiatric Clinics of North America 13(3):623–640, viii.
Dineen, R., P. M. Stewart, and M. Sherlock. 2019. Factors impacting on the action of glucocorticoids in patients receiving glucocorticoid therapy. Clinical Endocrinology 90(1):3–14.
Ding, Y., Y. Wang, Y. Lyu, H. Xie, Y. Huang, M. Wu, F. Chen, and Z. Chen. 2023. Urogenital sinus malformation: From development to management. Intractable & Rare Diseases Research 12(2):78–87.
Domenice, S., R. L. Batista, I. J. P. Arnhold, M. H. Sircili, E. M. F. Costa, and B. B. Mendonca. 2022. 46,XY differences of sexual development. In Endotext [Internet] edited by K. R. Feingold, B. Anawalt, M. R. Blackman, A. Boyce, G. Chrousos, E. Corpas, W. W. de Herder, K. Dhatariya, K. Dungan, J. Hofland, S. Kalra, G. Kaltsas, N. Kapoor, C. Koch, P. Kopp, M. Korbonits, C. S. Kovacs, W. Kuohung, B. Laferrère, M. Levy, E. A. McGee, R. McLachlan, M. New, J. Purnell, R. Sahay, A. S. Shah, F. Singer, M. A. Sperling, C. A. Stratakis, D. L. Trence, and D. P. Wilson. South Dartmouth, MA.
Duguid, A., S. Morrison, A. Robertson, J. Chalmers, G. Youngson, S. F. Ahmed, and the Scottish Genital Anomaly Network. 2007. The psychological impact of genital anomalies on the parents of affected children. Acta Paediatrica 96(3):348–352.
Dwyer, A. A., N. Smith, and R. Quinton. 2019. Psychological aspects of congenital hypogonadotropic hypogonadism. Frontiers in Endocrinology 10:353.
Edey, M. M. 2017. Male sexual dysfunction and chronic kidney disease. Frontiers in Medicine (Lausanne) 4:32.
El-Dahtory, F., and H. M. Elsheikha. 2009. Male infertility related to an aberrant karyotype, 47,XYY: Four case reports. Cases Journal 2(1):28.
Engberg, H., A. Strandqvist, A. Nordenstrom, A. Butwicka, A. Nordenskjold, A. L. Hirschberg, and L. Frisen. 2017. Increased psychiatric morbidity in women with complete androgen insensitivity syndrome or complete gonadal dysgenesis. Journal of Psychosomatic Research 101:122–127.
Ernst, M. M., L. M. Liao, A. B. Baratz, and D. E. Sandberg. 2018. Disorders of sex development/intersex: Gaps in psychosocial care for children. Pediatrics 142(2):e20174045.
Espinosa Reyes, T. M., A. K. Pesantez Velepucha, J. O. Cabrera Rego, W. Valdes Gomez, E. Dominguez Alonso, and H. Falhammar. 2023. Cardiovascular risk in Cuban adolescents and young adults with congenital adrenal hyperplasia. BMC Endocrine Disorders 23(1):241.
Falhammar, H., H. Filipsson Nyström, A. Wedell, and M. Thorén. 2011. Cardiovascular risk, metabolic profile, and body composition in adult males with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. European Journal of Endocrinology 164(2):285–293.
Falhammar, H., L. Frisen, C. Norrby, A. L. Hirschberg, C. Almqvist, A. Nordenskjold, and A. Nordenstrom. 2014. Increased mortality in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Endocrinology & Metabolism 99(12):e2715–2721.
Falhammar, H., H. Claahsen-van der Grinten, N. Reisch, J. Slowikowska-Hilczer, A. Nordenstrom, R. Roehle, C. Bouvattier, B. P. C. Kreukels, B. Kohler, and on behalf of the DSD-LIFE Group. 2018. Health status in 1040 adults with disorders of sex development (DSD): A European multicenter study. Endocrine Connections 7(3):466–478.
Falhammar, H., L. Frisen, A. L. Hirschberg, A. Nordenskjold, C. Almqvist, and A. Nordenstrom. 2022. Increased prevalence of fractures in congenital adrenal hyperplasia: A Swedish population-based national cohort study. Journal of Clinical Endocrinology & Metabolism 107(2):e475–e486.
Fang, J., G. Gao, J. Liu, L. Cai, Y. Cui, and X. Yang. 2021. A novel mutation of AMHR2 in two brothers with persistent Mullerian duct syndrome and their intracytoplasmic sperm injection outcome. Molecular Genetics & Genomic Medicine 9(10):e1801.
Farrugia, M. K., N. J. Sebire, J. C. Achermann, A. Eisawi, P. G. Duffy, and I. Mushtaq. 2013. Clinical and gonadal features and early surgical management of 45,X/46,XY and 45,X/47,XYY chromosomal mosaicism presenting with genital anomalies. Journal of Pediatric Urology 9(2):139–144.
Fleming, L., K. Knafl, G. Knafl, and M. Van Riper. 2017. Parental management of adrenal crisis in children with congenital adrenal hyperplasia. Journal for Specialists in Pediatric Nursing 22(4):e12190.
Foland-Ross, L. C., E. Ghasemi, V. Lozano Wun, T. Aye, K. Kowal, J. Ross, and A. L. Reiss. 2023. Executive dysfunction in Klinefelter syndrome: Associations with brain activation and testicular failure. Journal of Clinical Endocrinology & Metabolism 109(1):e88–e95.
Frank, S. E. 2018. Intersex and intimacy: Presenting concerns about dating and intimate relationships. Sexuality & Culture 22(1):127–147.
Furtado, P. S., F. Moraes, R. Lago, L. O. Barros, M. B. Toralles, and U. Barroso, Jr. 2012. Gender dysphoria associated with disorders of sex development. Nature Reviews Urology 9(11):620–627.
Gaston, B., N. Marozkina, D. C. Newcomb, N. Sharifi, and J. Zein. 2021. Asthma risk among individuals with androgen receptor deficiency. JAMA Pediatrics 175(7):743–745.
Ghazal, K., S. Brabant, D. Price, and M. L. Piketty. 2022. Hormone immunoassay interference: A 2021 update. Annals of Laboratory Medicine 42(1):3–23.
Gottlieb, B., L. K. Beitel, A. Nadarajah, M. Paliouras, and M. Trifiro. 2012. The androgen receptor gene mutations database: 2012 update. Human Mutation 33(5):887–894.
Granero-Molina, J., R. A. Roman, M. Del Mar Jimenez-Lasserrotte, M. D. Ruiz-Fernandez, M. I. Ventura-Miranda, G. Granero-Heredia, and I. M. Fernandez-Medina. 2023. “I’m still a woman”: A qualitative study on sexuality in heterosexual women with Turner syndrome. Journal of Clinical Nursing 32(17-18):6634–6647.
Grant, A., C. P. Carpenter, B. Li, and S. J. Kim. 2023. Hydrometrocolpos: A contemporary review of the last 5 years. Current Urology Reports 24(12):601–610.
Gravholt, C. H., A. Ferlin, J. Gromoll, A. Juul, A. Raznahan, S. van Rijn, A. D. Rogol, A. Skakkebaek, N. Tartaglia, and H. Swaab. 2023a. New developments and future trajectories in supernumerary sex chromosome abnormalities: A summary of the 2022 3rd International Workshop on Klinefelter Syndrome, Trisomy X, and XYY. Endocrine Connections 12(3):e220500.
Gravholt, C. H., M. Viuff, J. Just, K. Sandahl, S. Brun, J. van der Velden, N. H. Andersen, and A. Skakkebaek. 2023b. The changing face of Turner syndrome. Endocrine Reviews 44(1):33–69.
Gregg, A. R., B. G. Skotko, J. L. Benkendorf, K. G. Monaghan, K. Bajaj, R. G. Best, S. Klugman, and M. S. Watson. 2016. Noninvasive prenatal screening for fetal aneuploidy, 2016 update: A position statement of the American College of Medical Genetics and Genomics. Genetics in Medicine 18(10):1056–1065.
Grinspon, R. P., and R. A. Rey. 2019. Molecular characterization of XX maleness. International Journal of Molecular Sciences 20(23):6089.
Grinspon, R. P., S. Castro, and R. A. Rey. 2023. Up-to-date clinical and biochemical workup of the child and the adolescent with a suspected disorder of sex development. Hormone Research in Paediatrics 96(2):116–127.
Gürbüz, F., M. Alkan, G. Çelik, A. Bişgin, N. Çekin, I. Ünal, A. K. Topaloğlu, Ü. Zorludemir, A. Avcı, and B. Yüksel. 2020. Gender identity and assignment recommendations in disorders of sex development patients: 20 years’ experience and challenges. Journal of Clinical Research in Pediatric Endocrinology 12(4):347–357.
Haghighat, D., T. Berro, L. Torrey Sosa, K. Horowitz, B. Brown-King, and K. Zayhowski. 2023. Intersex people’s perspectives on affirming healthcare practices: A qualitative study. Social Science & Medicine 329:e116047.
Halaseh, S. A., S. Halaseh, and M. Ashour. 2022. Hypospadias: A comprehensive review including its embryology, etiology and surgical techniques. Cureus 14(7):e27544.
Haney, N. M., C. C. Morrill, A. Haffar, C. Crigger, A. T. Gabrielson, L. Galansky, and J. P. Gearhart. 2024. Long-term management of problems in cloacal exstrophy: A single-institution review. Journal of Pediatric Surgery 59(1):26–30.
Hannah-Shmouni, F., R. Morissette, N. Sinaii, M. Elman, T. R. Prezant, W. Chen, A. Pulver, and D. P. Merke. 2017. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in U.S. Ashkenazi Jews and Caucasians. Genetics in Medicine 19(11):1276–1279.
Herlin, M. K., M. B. Petersen, and M. Brannstrom. 2020. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: A comprehensive update. Orphanet Journal of Rare Diseases 15(1):214.
Herman, J. L., A. R. Flores, and K. K. O’Neill. 2022. How many adults and youth identify as transgender in the United States? Los Angeles, CA: The Williams Institute, UCLA School of Law.
Hines, M. 2020. Human gender development. Neuroscience Biobehavioral Reviews 118:89–96.
Howard, S. R., and L. Dunkel. 2018. Management of hypogonadism from birth to adolescence. Best Practice & Research Clinical Endocrinology & Metabolism 32(4):355–372.
Howard, S. R., and R. Quinton. 2024. Outcomes and experiences of adults with congenital hypogonadism can inform improvements in the management of delayed puberty. Journal of Pediatric Endocrinology & Metabolism 37(1):1–7.
Hsu, L. Y. 1989. Prenatal diagnosis of 45X/46,XY mosaicism: A review and update. Prenatal Diagnosis 9(1):31–48.
Hughes, I. A. 2007. Early management and gender assignment in disorders of sexual differentiation. Endocrine Development 11:47–57.
Hughes, I. A., C. Houk, S. F. Ahmed, P. A. Lee, L. C. Group, and the LWPES/ESPE Consensus Group. 2006. Consensus statement on management of intersex disorders. Archives of Disease in Childhood 91(7):554–563.
Hutaff-Lee, C., E. Bennett, S. Howell, and N. Tartaglia. 2019. Clinical developmental, neuropsychological, and social-emotional features of Turner syndrome. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 181(1):126–134.
Hwang, I. T. 2014. Efficacy and safety of growth hormone treatment for children born small for gestational age. Korean Journal of Pediatrics 57(9):379–383.
Ikegawa, K., and Y. Hasegawa. 2022. Fracture risk, underlying pathophysiology, and bone quality assessment in patients with Turner syndrome. Frontiers in Endocrinology 13:e967857.
Imperato-McGinley, J., M. Miller, J. D. Wilson, R. E. Peterson, C. Shackleton, and D. C. Gajdusek. 1991. A cluster of male pseudohermaphrodites with 5 alpha-reductase deficiency in Papua New Guinea. Clinical Endocrinology 34(4):293–298.
Izumita, Y., S. Nishigaki, M. Satoh, N. Takubo, C. Numakura, I. Takahashi, S. Soneda, Y. Abe, H. Kamasaki, Y. Ohtsu, J. Igaki, Y. Hasegawa, and K. Nagasaki. 2020. Retrospective study of the renal function using estimated glomerular filtration rate and congenital anomalies of the kidney-urinary tract in pediatric Turner syndrome. Congenital Anomalies 60(6):175–179.
Jackson, C., F. M. Cheater, and I. Reid. 2008. A systematic review of decision support needs of parents making child health decisions. Health Expectations 11(3):232–251.
Johannesson, M., C. Gottlieb, and L. Hjelte. 1997. Delayed puberty in girls with cystic fibrosis despite good clinical status. Pediatrics 99(1):29–34.
Jones, C. 2020. Intersex, infertility and the future: Early diagnoses and the imagined life course. Sociology of Health & Illness 42(1):143–156.
Jordan, T. L., L. C. Foland-Ross, V. L. Wun, J. L. Ross, and A. L. Reiss. 2023. Cognition, academic achievement, adaptive behavior, and quality of life in child and adolescent boys with Klinefelter syndrome. Journal of Developmental & Behavioral Pediatrics 44(7):e476–e485.
Kanakis, G. A., and E. Nieschlag. 2018. Klinefelter syndrome: More than hypogonadism. Metabolism 86:135–144.
Keppler-Noreuil, K. M., K. M. Conway, D. Shen, A. J. Rhoads, J. C. Carey, P. A. Romitti, and National Birth Defects Prevention Study. 2017. Clinical and risk factor analysis of cloacal defects in the national birth defects prevention study. American Journal of Medical Genetics Part A 173(11):2873–2885.
Kerckhof, M. E., B. P. C. Kreukels, T. O. Nieder, I. Becker-Hebly, T. C. van de Grift, A. S. Staphorsius, A. Kohler, G. Heylens, and E. Elaut. 2019. Prevalence of sexual dysfunctions in transgender persons: Results from the ENIGI follow-up study. Journal of Sexual Medicine 16(12):2018–2029.
Klein, D. A., J. E. Emerick, J. E. Sylvester, and K. S. Vogt. 2017. Disorders of puberty: An approach to diagnosis and management. American Family Physician 96(9):590–599.
Ko, J. K. Y., T. F. J. King, L. Williams, S. M. Creighton, and G. S. Conway. 2017. Hormone replacement treatment choices in complete androgen insensitivity syndrome: An audit of an adult clinic. Endocrine Connections 6(6):375–379.
Kohler, B., E. Kleinemeier, A. Lux, O. Hiort, A. Gruters, U. Thyen, and the DSD Network Working Group. 2012. Satisfaction with genital surgery and sexual life of adults with XY disorders of sex development: Results from the German clinical evaluation study. Journal of Clinical Endocrinology & Metabolism 97(2):577–588.
Kremen, J., S. M. Davis, L. Nahata, H. M. Kapa, T. M. Dattilo, E. Liu, C. Hutaff-Lee, A. C. Tishelman, and C. E. Crerand. 2023. Neuropsychological and mental health concerns in a multicenter clinical sample of youth with Turner syndrome. American Journal of Medical Genetics Part A 191(4):962–976.
Kreukels, B. P. C., B. Kohler, A. Nordenstrom, R. Roehle, U. Thyen, C. Bouvattier, A. L. C. de Vries, P. T. Cohen-Kettenis. 2018. Gender dysphoria and gender change in disorders of sex development/intersex conditions: Results from the DSD-life study. Journal of Sexual Medicine 15(5):777–785.
Lampalzer, U., P. Briken, and K. Schweizer. 2021. Psychosocial care and support in the field of intersex/diverse sex development (DSD): Counselling experiences, localisation and needed improvements. International Journal of Impotence Research 33(2):228–242.
Lee, P. A., C. P. Houk, S. Faisal Ahmed, I. A. Hughes, and the International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. 2006. Consensus statement on management of intersex disorders: International consensus conference on intersex. Pediatrics 118(2):e488–e500.
Lee, P. A., A. Nordenstrom, C. P. Houk, S. F. Ahmed, R. Auchus, A. Baratz, K. Baratz Dalke, L. M. Liao, K. Lin-Su, L. H. Looijenga, 3rd, T. Mazur, H. F. Meyer-Bahlburg, P. Mouriquand, C. A. Quigley, D. E. Sandberg, E. Vilain, S. Witchel, and the Global DSD Update Consortium. 2016. Global disorders of sex development update since 2006: Perceptions, approach and care. Hormone Research in Paediatrics 85(3):158–180.
Lindhardt Johansen, M., C. P. Hagen, E. Rajpert-De Meyts, S. Kjaergaard, B. L. Petersen, N. E. Skakkebaek, K. M. Main, and A. Juul. 2012. 45,X/46,XY mosaicism: Phenotypic characteristics, growth, and reproductive function: A retrospective longitudinal study. Journal of Clinical Endocrinology & Metabolism 97(8):e1540–e1549.
Lipstein, E. A., C. M. Dodds, and M. T. Britto. 2014. Real life clinic visits do not match the ideals of shared decision making. Journal of Pediatrics 165(1):178–183.E1.
Lucas-Herald, A. K., J. Bryce, A. Kyriakou, M. L. Ljubicic, W. Arlt, L. Audi, A. Balsamo, F. Baronio, S. Bertelloni, M. Bettendorf, A. Brooke, H. L. Claahsen van der Grinten, J. H. Davies, G. Hermann, L. de Vries, I. A. Hughes, R. Tadokoro-Cuccaro, F. Darendeliler, S. Poyrazoglu, M. Ellaithi, O. Evliyaoglu, S. Fica, L. Nedelea, A. Gawlik, E. Globa, N. Zelinska, T. Guran, A. Guven, S. E. Hannema, O. Hiort, P. M. Holterhus, V. Iotova, V. Mladenov, V. Jain, R. Sharma, F. Jennane, C. Johnston, G. Guerra Junior, D. Konrad, O. Gaisl, N. Krone, R. Krone, K. Lachlan, D. Li, C. Lichiardopol, L. Lisa, R. Markosyan, I. Mazen, K. Mohnike, M. Niedziela, A. Nordenstrom, R. Rey, M. Skaeil, L. J. W. Tack, J. Tomlinson, N. Weintrob, M. Cools, and S. F. Ahmed. 2021. Gonadectomy in conditions affecting sex development: A registry-based cohort study. European Journal of Endocrinology 184(6):791–801.
Lundberg, T., P. Hegarty, and K. Roen. 2018. Making sense of ‘intersex’ and ‘DSD’: How laypeople understand and use terminology. Psychology & Sexuality 9(2):161–173.
MacKenzie, D., A. Huntington, and J. A. Gilmour. 2009. The experiences of people with an intersex condition: A journey from silence to voice. Journal of Clinical Nursing 18(12):1775–1783.
Maimoun, L., P. Philibert, B. Cammas, F. Audran, P. Bouchard, P. Fenichel, M. Cartigny, C. Pienkowski, M. Polak, N. Skordis, I. Mazen, G. Ocal, M. Berberoglu, R. Reynaud, C. Baumann, S. Cabrol, D. Simon, K. Kayemba-Kay’s, M. De Kerdanet, F. Kurtz, B. Leheup, C. Heinrichs, S. Tenoutasse, G. Van Vliet, A. Grüters, M. Eunice, A. C. Ammini, M. Hafez, Z. Hochberg, S. Einaudi, H. Al Mawlawi, C. J. Nuñez, N. Servant, A. Lumbroso, F. Paris, and C. Sultan. 2011. Phenotypical, biological, and molecular heterogeneity of 5α-reductase deficiency: An extensive international experience of 55 patients. Journal of Clinical Endocrinology & Metabolism 96(2):296–307.
Marks, L. S. 2004. 5alpha-reductase: History and clinical importance. Reviews in Urology 6(Suppl 9):S11–S21.
Martínez-Frías, M. L., E. Bermejo, E. Rodriguez-Pinilla, and J. L. Frias. 2001. Exstrophy of the cloaca and exstrophy of the bladder: Two different expressions of a primary developmental field defect. American Journal of Medical Genetics 99(4):261–269.
Maruf, M., R. Manyevitch, J. Michaud, J. Jayman, M. Kasprenski, M. H. Zaman, K. Benz, M. Eldridge, B. Trock, K. T. Harris, W. J. Wu, H. N. Di Carlo, and J. P. Gearhart. 2020. Urinary continence outcomes in classic bladder exstrophy: A long-term perspective. Journal of Urology 203(1):200–205.
Mayo Clinic. 2018. Ambiguous genitalia. https://www.mayoclinic.org/diseases-conditions/ambiguous-genitalia/diagnosis-treatment/drc-20369278 (accessed March 13, 2024).
Mazur, T., J. O’Donnell, and P. A. Lee. 2023. Extensive literature review of 46,XX newborns with congenital adrenal hyperplasia (CAH) and severe genital masculinization: Should they be assigned and reared male? Journal of Research in Pediatric Endocrinology 16(2):123–136.
McCarrison, S., A. Carr, S. C. Wong, and A. Mason. 2023. The prevalence of hypertension in paediatric Turner syndrome: A systematic review and meta-analysis. Journal of Human Hypertension 37(8):675–688.
McCoy, R. C. 2017. Mosaicism in preimplantation human embryos: When chromosomal abnormalities are the norm. Trends in Genetics 33(7):448–463.
Meccanici, F., J. W. C. de Bruijn, J. S. Dommisse, J. J. M. Takkenberg, A. E. van den Bosch, and J. W. Roos-Hesselink. 2023. Prevalence and development of aortic dilation and dissection in women with Turner syndrome: A systematic review and meta-analysis. Expert Review of Cardiovascular Therapy 21(2):133–144.
Mendoza, N., and M. A. Motos. 2013. Androgen insensitivity syndrome. Gynecological Endocrinology 29(1):1–5.
Meyer-Bahlburg, H. F., R. S. Gruen, M. I. New, J. J. Bell, A. Morishima, M. Shimshi, Y. Bueno, I. Vargas, and S. W. Baker. 1996. Gender change from female to male in classical congenital adrenal hyperplasia. Hormones and Behavior 30(4):319–332.
Miller, W. L., and R. J. Auchus. 2011. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Reviews 32(1):81–151.
Mitsch, C., E. Alexandrou, A. W. Norris, and C. T. Pinnaro. 2023. Hyperglycemia in Turner syndrome: Impact, mechanisms, and areas for future research. Frontiers in Endocrinology 14:e1116889.
Mittal, S., J. Weaver, A. Aghababian, R. Edwins, K. Godlewski, K. Fischer, S. Siu, D. Gruccio, J. Van Batavia, A. Srinivasan, C. Long, V. Bamba, V. Batra, T. Bhatti, and T. Kolon. 2023. Deferring gonadectomy in patients with Turner syndrome with a genetic Y component is not a safe practice. Journal of Pediatric Urology 19(3):e291–e294.
Momodu, I. I., B. Lee, and G. Singh. 2023. Congenital adrenal hyperplasia. [Updated 2023 July 17], Statpearls. Treasure Island, FL: StatPearls Publishing.
Money, J. 1952. Hermaphroditism: An inquiry into the nature of a human paradox. Doctoral Thesis, Harvard University.
Moran, M. E., and K. Karkazis. 2012. Developing a multidisciplinary team for disorders of sex development: Planning, implementation, and operation tools for care providers. Advances in Urology 2012:e604135.
Moreno-Begines, M. L. N., A. Arroyo-Rodriguez, A. Borrallo-Riego, and M. D. Guerra-Martin. 2022. Intersexuality/differences of sex development through the discourse of intersex people, their relatives, and health experts: A descriptive qualitative study. Healthcare (Basel, Switzerland) 10(4):671.
Morris, J. K., E. Alberman, C. Scott, and P. Jacobs. 2008. Is the prevalence of Klinefelter syndrome increasing? European Journal of Human Genetics 16(2):163–170.
Naessén, S., M. Eliasson, K. Berntorp, M. Kitlinski, P. Trimpou, E. Amundson, S. Thunstrom, B. Ekman, J. Wahlberg, A. Karlsson, M. Isaksson, I. Bergstrom, C. Levelind, I. Bryman, and K. Landin-Wilhelmsen. 2024. Autoimmune disease in Turner syndrome in Sweden: An up to 25 years’ controlled follow-up study. Journal of Clinical Endocrinology & Metabolism 109(2):e602–e612.
Neel, N., and M. S. Tarabay. 2018. Omphalocele, exstrophy of cloaca, imperforate anus, and spinal defect complex, multiple major reconstructive surgeries needed. Urology Annals 10(1):118–121.
Ng, S. M., K. M. Stepien, and A. Krishan. 2020. Glucocorticoid replacement regimens for treating congenital adrenal hyperplasia. Cochrane Database of Systematic Reviews 3(3):CD012517.
Ogawa, T., F. Takizawa, Y. Mukoyama, A. Ogawa, and J. Ito. 2021. Renal morphology and function from childhood to adulthood in Turner syndrome. Clinical and Experimental Nephrology 25(6):633–640.
Palmert, M. R., and L. Dunkel. 2012. Clinical practice: Delayed puberty. New England Journal of Medicine 366(5):443–453.
Pasterski, V., P. Prentice, and I. A. Hughes. 2010. Impact of the consensus statement and the new DSD classification system. Best Practice & Research Clinical Endocrinology & Metabolism 24(2):187–195.
Pasterski, V., K. Mastroyannopoulou, D. Wright, K. J. Zucker, and I. A. Hughes. 2014. Predictors of posttraumatic stress in parents of children diagnosed with a disorder of sex development. Archives of Sexual Behavior 43(2):369–375.
Pasterski, V., K. J. Zucker, P. C. Hindmarsh, I. A. Hughes, C. Acerini, D. Spencer, S. Neufeld, and M. Hines. 2015. Increased cross-gender identification independent of gender role behavior in girls with congenital adrenal hyperplasia: Results from a standardized assessment of 4- to 11-year-old children. Archives of Sexual Behavior 44(5):1363–1375.
Persani, L., M. Cools, S. Ioakim, S. Faisal Ahmed, S. Andonova, M. Avbelj-Stefanija, F. Baronio, J. Bouligand, H. T. Bruggenwirth, J. H. Davies, E. De Baere, I. Dzivite-Krisane, P. Fernandez-Alvarez, A. Gheldof, C. Giavoli, C. H. Gravholt, O. Hiort, P. M. Holterhus, A. Juul, C. Krausz, K. Lagerstedt-Robinson, R. McGowan, U. Neumann, A. Novelli, X. Peyrassol, L. A. Phylactou, J. Rohayem, P. Touraine, D. Westra, V. Vezzoli, and R. Rossetti. 2022. The genetic diagnosis of rare endocrine disorders of sex development and maturation: A survey among endo-ERN centres. Endocrine Connections 11(12):e220367.
Pofi, R., X. Ji, N. P. Krone, and J. W. Tomlinson. 2023. Long-term health consequences of congenital adrenal hyperplasia. Clinical Endocrinology. Advanced online publication. https://doi.org/10.1111/cen.14967.
Porsius, E., M. Spath, K. Kluivers, W. Klein, and H. Claahsen-van der Grinten. 2022. Primary amenorrhea with apparently absent uterus: A report of three cases. Journal of Clinical Medicine 11(15):4305.
Puar, T. H., N. M. Stikkelbroeck, L. C. Smans, P. M. Zelissen, and A. R. Hermus. 2016. Adrenal crisis: Still a deadly event in the 21st century. American Journal of Medicine 129(3):e331–e339.
Ranke, M. B. 1995. Growth hormone therapy in Turner syndrome: Analysis of long-term results. Hormone Research 44(Suppl 3):35–41.
Reiner, W. G., and J. P. Gearhart. 2004. Discordant sexual identity in some genetic males with cloacal exstrophy assigned to female sex at birth. New England Journal of Medicine 350(4):333–341.
Rhodes, M., S. A. Akohoue, S. M. Shankar, I. Fleming, A. Q. An, C. Yu, S. Acra, and M. S. Buchowski. 2009. Growth patterns in children with sickle cell anemia during puberty. Pediatric Blood & Cancer, 53(4):635–641.
Richards, S., N. Aziz, S. Bale, D. Bick, S. Das, J. Gastier-Foster, W. W. Grody, M. Hegde, E. Lyon, E. Spector, K. Voelkerding, H. L. Rehm, and the ACMG Laboratory Quality Assurance Committee. 2015. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 17(5):405–424.
Ridder, L. O., A. Berglund, K. Stochholm, S. Chang, and C. H. Gravholt. 2023. Morbidity, mortality, and socioeconomics in Klinefelter syndrome and 47,XYY syndrome: A comparative review. Endocrine Connections 12(5):e230024.
Righi, B., S. R. Ali, J. Bryce, J. W. Tomlinson, W. Bonfig, F. Baronio, E. C. Costa, G. GuaragnaFilho, G. T’Sjoen, M. Cools, R. Markosyan, T. Bachega, M. C. Miranda, V. Iotova, H. Falhammar, F. Ceccato, M. R. Stancampiano, G. Russo, E. Daniel, R. J. Auchus, R. J. Ross, and S. F. Ahmed. 2023. Long-term cardiometabolic morbidity in young adults with classic 21-hydroxylase deficiency congenital adrenal hyperplasia. Endocrine 80(3):630–638.
Rodriguez-Wallberg, K. A., F. Sergouniotis, H. P. Nilsson, and F. E. Lundberg. 2023. Trends and outcomes of fertility preservation for girls, adolescents and young adults with Turner syndrome: A prospective cohort study. Frontiers in Endocrinology 14:e1135249.
Romejko, K., A. Rymarz, H. Sadownik, and S. Niemczyk. 2022. Testosterone deficiency as one of the major endocrine disorders in chronic kidney disease. Nutrients 14(16):3438.
Rosenwohl-Mack, A., S. Tamar-Mattis, A. B. Baratz, K. B. Dalke, A. Ittelson, K. Zieselman, and J. D. Flatt. 2020. A national study on the physical and mental health of intersex adults in the U.S. PLoS ONE [Electronic Resource] 15(10):e0240088.
Ross, J. L., L. M. Long, M. Skerda, F. Cassorla, D. Kurtz, D. L. Loriaux, and G. B. Cutler, Jr. 1986. Effect of low doses of estradiol on 6-month growth rates and predicted height in patients with Turner syndrome. Journal of Pediatrics 109(6):950–953.
Saari, A., S. Harju, O. Makitie, M. T. Saha, L. Dunkel, and U. Sankilampi. 2015. Systematic growth monitoring for the early detection of celiac disease in children. JAMA Pediatrics 169(3):e1525.
Saggese, G., S. Bertelloni, and G. I. Baroncelli. 1997. Sex steroids and the acquisition of bone mass. Hormone Research 48(Suppl 5):65–71.
Samarasinghe, S., F. Meah, V. Singh, A. Basit, N. Emanuele, M. A. Emanuele, A. Mazhari, and E. W. Holmes. 2017. Biotin interference with routine clinical immunoassays: Understand the causes and mitigate the risks. Endocrine Practice 23(8):989–998.
Sandahl, K., J. Wen, M. Erlandsen, N. H. Andersen, and C. H. Gravholt. 2020. Natural history of hypertension in Turner syndrome during a 12-year pragmatic interventional study. Hypertension 76(5):1608–1615.
Sandberg, D. E., M. Gardner, and P. T. Cohen-Kettenis. 2012. Psychological aspects of the treatment of patients with disorders of sex development. Seminars in Reproductive Medicine 30(5):443–452.
Sandberg, D. E., M. Gardner, N. Callens, T. Mazur, the DSD-TRN Psychosocial Workgroup, the DSD-TRN Advocacy Advisory Network, and the Accord Alliance. 2017. Interdisciplinary care in disorders/differences of sex development (DSD): The psychosocial component of the DSD-translational research network. American Journal of Medical Genetics Seminars in Medical Genetics Part C 175(2):279–292.
Sandberg, D. E., M. Gardner, K. Kopec, M. Urbanski, N. Callens, C. E. Keegan, B. M. Yashar, P. Y. Fechner, M. Shnorhavorian, E. Vilain, S. Timmermans, and L. A. Siminoff. 2019. Development of a decision support tool in pediatric differences/disorders of sex development. Seminars in Pediatric Surgery 28(5):150838.
Saulnier, K. M., H. Gallois, and Y. Joly. 2021. Prenatal genetic testing for intersex conditions in Canada. Journal of Obstetrics and Gynaecology Canada 43(3):369–371.
Schönbucher, V., K. Schweizer, L. Rustige, K. Schutzmann, F. Brunner, and H. Richter-Appelt. 2012. Sexual quality of life of individuals with 46,XY disorders of sex development. Journal of Sexual Medicine 9(12):3154–3170.
Schweizer, K., F. Brunner, K. Schützmann, V. Schönbucher, and H. Richter-Appelt. 2009. Gender identity and coping in female 46,XY adults with androgen biosynthesis deficiency (intersexuality/DSD). Journal of Counseling Psychology 56(1):189–201.
Seminog, O. O., A. B. Seminog, D. Yeates, and M. J. Goldacre. 2015. Associations between Klinefelter’s syndrome and autoimmune diseases: English national record linkage studies. Autoimmunity 48(2):125–128.
Sengupta, P., S. Dutta, I. R. Karkada, and S. V. Chinni. 2021. Endocrinopathies and male infertility. Life 12(1):10.
Speiser, P. W., W. Arlt, R. J. Auchus, L. S. Baskin, G. S. Conway, D. P. Merke, H. F. L. Meyer-Bahlburg, W. L. Miller, M. H. Murad, S. E. Oberfield, and P. C. White. 2018. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. Journal of Clinical Endocrinology & Metabolism 103(11):4043–4088.
SSA (Social Security Administration). 2017. Revisions to rules regarding the evaluation of medical evidence. Federal Register 82(5869).
Stancampiano, M. R., K. Suzuki, S. O’Toole, G. Russo, G. Yamada, and S. Faisal Ahmed. 2022. Congenital micropenis: Etiology and management. Journal of the Endocrine Society 6(2):bvab172.
Sullivan, S. D., P. M. Sarrel, and L. M. Nelson. 2016. Hormone replacement therapy in young women with primary ovarian insufficiency and early menopause. Fertility and Sterility 106(7):1588–1599.
Sultan, C., P. Philibert, L. Gaspari, F. Audran, L. Maimoun, N. Kalfa, and F. Paris. 2014. Chapter 5 - androgen insensitivity syndrome. In Genetic steroid disorders, edited by M. I. New, O. Lekarev, A. Parsa, T. T. Yuen, B. W. O’Malley and G. D. Hammer. San Diego: Academic Press. Pp. 225–237.
Sutton, E. J., A. McInerney-Leo, C. A. Bondy, S. E. Gollust, D. King, and B. Biesecker. 2005. Turner syndrome: Four challenges across the lifespan. American Journal of Medical Genetics Part A 139A(2):57–66.
Tack, L. J. W., E. Maris, L. H. J. Looijenga, S. E. Hannema, L. Audi, B. Kohler, P. M. Holterhus, S. Riedl, A. Wisniewski, C. E. Fluck, J. H. Davies, G. T’Sjoen, A. K. Lucas-Herald, O. Evliyaoglu, N. Krone, V. Iotova, O. Marginean, A. Balsamo, G. Verkauskas, N. Weintrob, M. Ellaithi, A. Nordenstrom, A. Verrijn Stuart, K. B. Kluivers, K. P. Wolffenbuttel, S. F. Ahmed, and M. Cools. 2018. Management of gonads in adults with androgen insensitivity: An international survey. Hormone Research in Paediatrics 90(4):236–246.
Tallaksen, H. B. L., E. B. Johannsen, J. Just, M. H. Viuff, C. H. Gravholt, and A. Skakkebaek. 2023. The multi-omic landscape of sex chromosome abnormalities: Current status and future directions. Endocrine Connections 12(9):e230011.
Tartaglia, N. R., R. Wilson, J. S. Miller, J. Rafalko, L. Cordeiro, S. Davis, D. Hessl, and J. Ross. 2017. Autism spectrum disorder in males with sex chromosome aneuploidy: XXY/Klinefelter syndrome, XYY, and XXYY. Journal of Developmental & Behavioral Pediatrics 38(3):197–207.
Therrell, B. L. 2001. Newborn screening for congenital adrenal hyperplasia. Endocrinology & Metabolism Clinics of North America 30(1):15–30.
Thigpen, A. E., D. L. Davis, A. Milatovich, B. B. Mendonca, J. Imperato-McGinley, J. E. Griffin, U. Francke, J. D. Wilson, and D. W. Russell. 1992. Molecular genetics of steroid 5 alpha-reductase 2 deficiency. Journal of Clinical Investigation 90(3):799–809.
Thunström S., E. Thunström, S. Naessén, K. Berntorp, M. Laczna Kitlinski, B. Ekman, J. Wahlberg, I. Bergström, O. Bech-Hanssen, E. Krantz, C. M. Laine, I. Bryman, and K. Landin-Wilhelmsen. 2023. Aortic size predicts aortic dissection in Turner syndrome: A 25-year prospective cohort study. International Journal of Cardiology 373: 47–54.
Thyen, U., A. Lux, M. Jurgensen, O. Hiort, and B. Kohler. 2014. Utilization of health care services and satisfaction with care in adults affected by disorders of sex development (DSD). Journal of General Internal Medicine 29(Suppl 3):S752–S759.
Tomaselli, S., F. Megiorni, C. De Bernardo, A. Felici, G. Marrocco, G. Maggiulli, B. Grammatico, D. Remotti, P. Saccucci, F. Valentini, M. C. Mazzilli, S. Majore, and P. Grammatico. 2008. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Human Mutation 29(2):220–226.
Tomlinson, C., H. Macintyre, C. A. Dorrian, S. F. Ahmed, and A. M. Wallace. 2004. Testosterone measurements in early infancy. Archives of Disease in Childhood: Fetal and Neonatal Edition 89(6):F558–F559.
Torky, A., N. Sinaii, S. Jha, J. Desai, D. El-Maouche, A. Mallappa, and D. P. Merke. 2021. Cardiovascular disease risk factors and metabolic morbidity in a longitudinal study of congenital adrenal hyperplasia. Journal of Clinical Endocrinology & Metabolism 106(12):e5247–e5257.
Traino, K. A., C. M. Roberts, R. S. Fisher, A. M. Delozier, P. F. Austin, L. S. Baskin, Y. M. Chan, E. Y. Cheng, D. A. Diamond, A. J. Fried, B. Kropp, Y. Lakshmanan, S. Z. Meyer, T. Meyer, C. Buchanan, B. W. Palmer, A. Paradis, K. J. Reyes, A. Tishelman, P. Williot, C. Wolfe-Christensen, E. B. Yerkes, L. L. Mullins, and A. B. Wisniewski. 2022. Stigma, intrusiveness, and distress in parents of children with a disorder/difference of sex development. Journal of Developmental & Behavioral Pediatrics 43(7):e473–e482.
T’Sjoen, G., G. De Cuypere, S. Monstrey, P. Hoebeke, F. K. Freedman, M. Appari, P. M. Holterhus, J. Van Borsel, and M. Cools. 2011. Male gender identity in complete androgen insensitivity syndrome. Archives of Sexual Behavior 40(3):635–638.
Tyutyusheva, N., I. Mancini, G. I. Baroncelli, S. D’Elios, D. Peroni, M. C. Meriggiola, and S. Bertelloni. 2021. Complete androgen insensitivity syndrome: From bench to bed. International Journal of Molecular Sciences 22(3):1264.
Urban, M. D., P. A. Lee, J. P. Dorst, L. P. Plotnick, and C. J. Migeon. 1979. Oxandrolone therapy in patients with Turner syndrome. Journal of Pediatrics 94(5):823–827.
van de Grift, T. C., M. Rapp, G. Holmdahl, L. Duranteau, A. Nordenskjold, and the DSD-LIFE Group. 2022. Masculinizing surgery in disorders/differences of sex development: Clinician- and participant-evaluated appearance and function. BJU International 129(3):394–405.
Van den Eede, E., M. Sterckx, K. Vangelabbeek, C. Dunford, A. Noah, D. Wood, and G. De Win. 2023. An observational study on the sexual, genital and fertility outcomes in bladder exstrophy and epispadias patients. Journal of Pediatric Urology 19(1):e31–e37.
van der Horst, H. J., and L. L. de Wall. 2017. Hypospadias: All there is to know. European Journal of Pediatrics 176(4):435–441.
van der Zwan, Y. G., N. Callens, J. van Kuppenveld, K. Kwak, S. L. Drop, B. Kortmann, A. B. Dessens, K. P. Wolffenbuttel, and the Dutch Study Group on DSD. 2013. Long-term outcomes in males with disorders of sex development. Journal of Urology 190(3):1038–1042.
van Lisdonk, J. 2014. Living with intersex/DSD: An exploratory study of the social situation of persons with intersex/DSD. The Hague, Netherlands: The Netherlands Institute for Social Research.
van Rijn, S., L. Stockmann, M. Borghgraef, H. Bruining, C. van Ravenswaaij-Arts, L. Govaerts, K. Hansson, and H. Swaab. 2014. The social behavioral phenotype in boys and girls with an extra X chromosome (Klinefelter syndrome and trisomy X): A comparison with autism spectrum disorder. Journal of Autism and Developmental Disorders 44(2):310–320.
Villanueva, C., and J. Argente. 2014. Pathology or normal variant: What constitutes a delay in puberty? Hormone Research in Paediatrics 82(4):213–221.
Weidler, E. M., and K. E. Peterson. 2019. The impact of culture on disclosure in differences of sex development. Seminars in Pediatric Surgery 28(5):e150840.
Weijenborg, P. T. M., K. B. Kluivers, A. B. Dessens, M. J. Kate-Booij, and S. Both. 2019. Sexual functioning, sexual esteem, genital self-image and psychological and relational functioning in women with Mayer-Rokitansky-Kuster-Hauser syndrome: A case-control study. Human Reproduction 34(9):1661–1673.
Wilkins, L., M. M. Grumbach, J. J. Van Wyk, T. H. Shepard, and C. Papadatos. 1955. Hermaphroditism: Classification, diagnosis, selection of sex and treatment. Pediatrics 16(3):287–302.
Wisniewski, A. B. 2012. Gender development in 46,XY DSD: Influences of chromosomes, hormones, and interactions with parents and healthcare professionals. Scientifica 2012:834967.
Wisniewski, A. B., R. L. Batista, E. M. F. Costa, C. Finlayson, M. H. P. Sircili, F. T. Denes, S. Domenice, and B. B. Mendonca. 2019. Management of 46,XY differences/disorders of sex development (DSD) throughout life. Endocrine Reviews 40(6):1547–1572.
Witchel, S. F., T. Mazur, C. P. Houk, and P. A. Lee. 2022. The long path to our current understanding regarding care of children with differences/disorders of sexual development. Hormone Research in Paediatrics 95(6):608–618.
Wu, Q. Y., N. Li, W. W. Li, T. F. Li, C. Zhang, Y. X. Cui, X. Y. Xia, and J. S. Zhai. 2014. Clinical, molecular and cytogenetic analysis of 46,XX testicular disorder of sex development with SRY-positive. BMC Urology 14:70.
Wudy, S. A., G. Schuler, A. Sanchez-Guijo, and M. F. Hartmann. 2018. The art of measuring steroids: Principles and practice of current hormonal steroid analysis. Journal of Steroid Biochemistry & Molecular Biology 179:88–103.
Yatsenko, S. A., and A. Rajkovic. 2019. Genetics of human female infertility. Biology of Reproduction 101(3):549–566.
Yoon, J. C., J. L. Casella, M. Litvin, and A. S. Dobs. 2019. Male reproductive health in cystic fibrosis. Journal of Cystic Fibrosis 18:S105–S110.
Yoon, S. H., G. Y. Kim, G. T. Choi, and J. T. Do. 2023. Organ abnormalities caused by Turner syndrome. Cells 12(10):1365.
Zahra, B., A. Sastry, M. Freel, M. Donaldson, and A. Mason. 2023. Turner syndrome transition clinic in the west of Scotland: A perspective. Frontiers in Endocrinology 14:e1233723.
Zucker, K. J., S. J. Bradley, G. Oliver, J. Blake, S. Fleming, and J. Hood. 1996. Psychosexual development of women with congenital adrenal hyperplasia. Hormones and Behavior 30(4):300–318.
This page intentionally left blank.





















































